




Содержание
Перейти к:
https://doi.org/10.30895/2312-7821-2024-12-3-331-340
Перейти к:
ВВЕДЕНИЕ. Документы о системе фармаконадзора держателя регистрационного удостоверения (ДРУ) — мастер-файл системы фармаконадзора (МФСФ), а также составляемая на его основе краткая характеристика системы фармаконадзора — являются обязательной частью регистрационного досье лекарственного препарата. Заявитель должен представлять и обновлять эти документы строго в соответствии с законодательством Евразийского экономического союза (ЕАЭС). Систематизация требований к оформлению и подаче документов, описывающих систему фармаконадзора, позволит заявителям оптимизировать процессы их подготовки.
ЦЕЛЬ. Анализ требований к представлению МФСФ и краткой характеристики системы фармаконадзора ДРУ в зависимости от регистрационных процедур ЕАЭС, описание типичных ошибок заявителей при представлении этих документов.
ОБСУЖДЕНИЕ. Содержание документов о системе фармаконадзора в составе регистрационного досье регулируется Правилами надлежащей практики фармаконадзора Евразийского экономического союза (Решение Совета Евразийской экономической комиссии (ЕЭК) от 03.11.2016 № 87), представление — Правилами регистрации и экспертизы лекарственных средств для медицинского применения (Решение Совета ЕЭК от 03.11.2016 № 78). ДРУ обязаны поддерживать в актуальном состоянии как сами документы, так и регистрационные досье, в которые они включены. В статье обобщены особенности предоставления МФСФ либо краткой характеристики системы фармаконадзора в зависимости от процедуры, заявленной на экспертизу, и типичные ошибки ДРУ при подготовке документов о системе фармаконадзора. При подаче заявления на регистрацию по Правилам надлежащей практики фармаконадзора ЕАЭС лекарственного препарата, который является первым для ДРУ на фармацевтическом рынке ЕАЭС, в регистрационное досье включают МФСФ. При последующих заявках на регистрацию лекарственных препаратов данного ДРУ в составе регистрационного досье подается краткая характеристика системы фармаконадзора. Изменения в документы по фармаконадзору вносятся в соответствии с классификатором (Решение Совета ЕЭК от 03.11.2016 № 78).
ВЫВОДЫ. Проведенный экспертами анализ требований к подаче МФСФ и краткой характеристики системы фармаконадзора ДРУ при различных регистрационных процедурах будет способствовать соблюдению заявителями требований законодательных актов ЕАЭС, корректному представлению необходимых документов о системе фармаконадзора и позволит уменьшить количество запросов со стороны регуляторных органов о представлении недостающей информации и отказов в регистрации лекарственного препарата.
ERRATUM
Исправление к статье Вельц Н.Ю. и соавт. «Представление документов о системе фармаконадзора в составе регистрационного досье в рамках процедур ЕАЭС: анализ требований и типичных ошибок»
https://www.risksafety.ru/jour/article/view/476
Вельц Н.Ю., Журавлева Е.О., Кутехова Г.В., Терешкина Н.В. Представление документов о системе фармаконадзора в составе регистрационного досье в рамках процедур ЕАЭС: анализ требований и типичных ошибок. Безопасность и риск фармакотерапии. 2024;12(3):331-340. https://doi.org/10.30895/2312-7821-2024-12-3-331-340
Velts N.Yu., Zhuravleva E.O., Kutekhova G.V., Tereshkina N.V. Submission of Documents on the Pharmacovigilance System as Part of the Registration Dossier within the Framework of the EAEU Procedures: Analysis of Requirements and Typical Errors. Safety and Risk of Pharmacotherapy. 2024;12(3):331-340. (In Russ.) https://doi.org/10.30895/2312-7821-2024-12-3-331-340
Система фармаконадзора (СФ) создается держателями регистрационных удостоверений (ДРУ) для выполнения функций и обязательств по фармаконадзору1. ДРУ совместно с уполномоченными органами в сфере фармаконадзора контролирует безопасность лекарственного препарата (ЛП), выявляя все изменения, влияющие на соотношение «польза–риск», что позволяет оперативно принимать необходимые меры для превышения пользы над риском. Эффективная СФ, организованная в соответствии с Правилами надлежащей практики фармаконадзора Евразийского экономического союза (ЕАЭС)2, обеспечивает проведение непрерывного мониторинга, своевременный сбор и анализ сообщений по безопасности ЛП, последующую обработку данных с выявлением новых сигналов и незамедлительную передачу информации по безопасности в регуляторные органы [1].
Для документального подтверждения соответствия СФ требованиям нормативных правовых актов в сфере обращения лекарственных средств в ЕАЭС3 ДРУ создает подробное описание СФ (ПОСФ), представленное в мастер-файле системы фармаконадзора (МФСФ) [2], и краткую характеристику системы фармаконадзора (КХСФ). Информация, содержащаяся в этих документах, позволяет осуществлять оценку организации и эффективности работы СФ в первую очередь самому ДРУ, в том числе при проведении аудитов, а также регуляторным органам при проведении экспертизы документов и осуществлении инспекций СФ [3][4].
Данные о безопасности лекарственного препарата имеют особое значение в регистрационном досье [5]. МФСФ и КХСФ позволяют оценить наличие и эффективность функционирования СФ ДРУ, а также необходимость проведения инспекции СФ. Эти документы необходимо своевременно актуализировать и предоставлять строго в соответствии с требованиями законодательства ЕАЭС.
Систематизация требований к оформлению и подаче документов, описывающих СФ, позволит заявителям оптимизировать процессы их подготовки.
Цель работы — анализ требований к представлению мастер-файла системы фармаконадзора и краткой характеристики системы фармаконадзора держателя регистрационного удостоверения в зависимости от регистрационных процедур Евразийского экономического союза, а также описание типичных ошибок заявителей при представлении этих документов.
Документы о СФ ДРУ входят в состав регистрационного досье (РД) ЛП при подаче заявления для получения регистрационного удостоверения на ЛП, содержащий как известное действующее вещество (комбинацию действующих веществ), так и новое действующее вещество (комбинацию действующих веществ) [6].
МФСФ подробно описывает СФ, применяемую ДРУ в отношении выпускаемых на фармацевтический рынок ЛП. Информация в МФСФ должна отражать действующую СФ в текущий момент времени, поэтому ДРУ необходимо своевременно ее обновлять и поддерживать в актуальном состоянии (п. 126 Правил надлежащей практики фармаконадзора ЕАЭС). Перечень разделов МФСФ и их наполнение должны соответствовать нормативным требованиям4 [2][7]. Заявителям важно принимать во внимание, что если при проведении экспертизы документов о СФ в РД выявляется их несоответствие указанным требованиям, экспертное учреждение направляет запрос к ДРУ о предоставлении недостающей информации и устранении недочетов. Окончательное заключение по СФ эксперт формирует после получения ответа на данный запрос.
ДРУ в соответствии с Правилами надлежащей практики фармаконадзора ЕАЭС включает МФСФ в РД при регистрации первого препарата на фармацевтическом рынке Союза. Далее при подаче документов на регистрацию ЛП тем же ДРУ в состав РД включается уже не МФСФ, а КХСФ5. КХСФ подтверждает, что СФ разработана и внедрена в работу компании, назначено и выполняет свои функции уполномоченное лицо по фармаконадзору (УЛФ), а также содержит указание на местонахождение МФСФ.
МФСФ размещают в разделе 1.10.1 модуля 1 РД в виде документа с кодом 07001 согласно классификатору видов документов РД ЛП6, утвержденного Решением Коллегии Евразийской экономической комиссии (ЕЭК) от 17.09.2019 № 159. КХСФ — в том же разделе 1.10.1 РД, но в виде документа с кодом 07002. В разделе 1.10.2 РД необходимо представить письменное подтверждение ДРУ факта наличия уполномоченного лица, ответственного за фармаконадзор на территории государств — членов ЕАЭС (рис. 1).
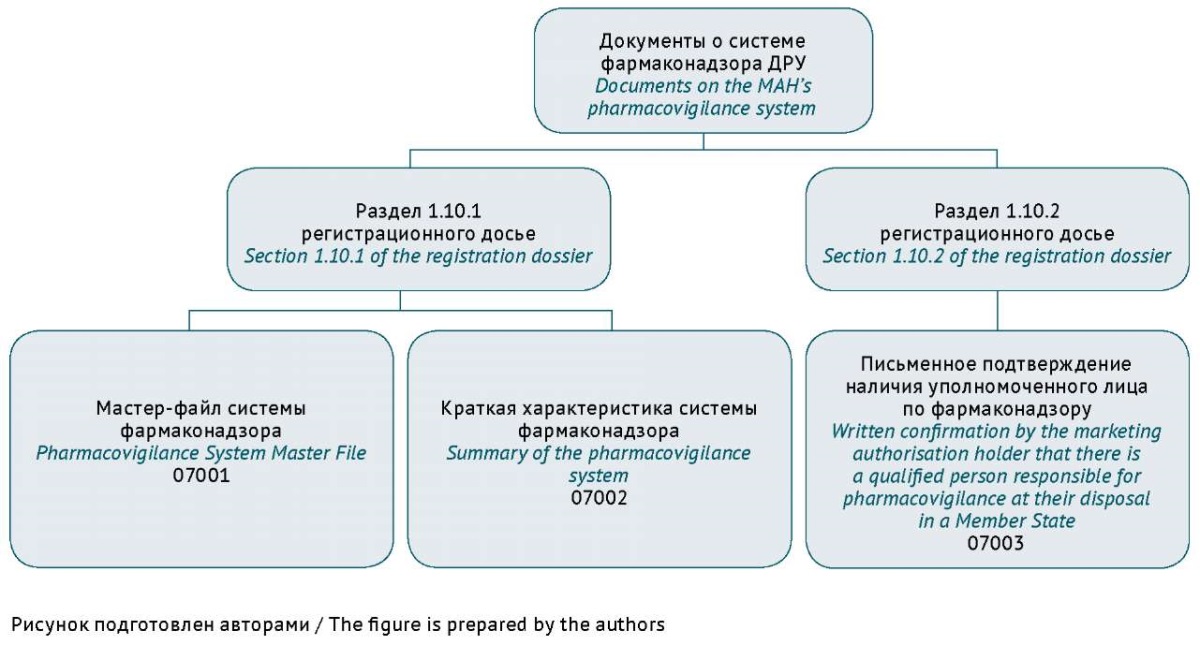
Рис. 1. Документы о системе фармаконадзора держателя регистрационного удостоверения (ДРУ) в составе регистрационного досье лекарственного препарата
Fig. 1. Documents on the pharmacovigilance system of the marketing authorisation holder (MAH) in the registration dossier of a medicinal product
В настоящее время единый реестр МФСФ в ЕАЭС отсутствует, поэтому при подаче КХСФ рекомендуется указывать ЛП, в РД которого находится актуальный МФСФ. Кроме того, при подаче документов следует убедиться, что описываемая СФ принадлежит ДРУ, указанному в заявлении на регистрацию и документации, представленной в РД (например, общей характеристике ЛП, инструкции по медицинскому применению, листке-вкладыше, нормативным документам по качеству и т.п.).
Порядок предоставления документов о СФ при заявлении ЛП на регистрацию четко определен в Решениях Совета ЕЭК7 [8]. Выделяют следующие процедуры регистрации:
Порядок предоставления документов является общим независимо от типа регистрации и указан в разделе статьи «Документы о системе фармаконадзора».
При инициировании процедуры приведения РД ЛП в соответствие с требованиями ЕАЭС в разделе 1.10.1 модуля 1 РД в обязательном порядке должна быть представлена информация о СФ ДРУ (Приложение № 1 к Правилам регистрации и экспертизы ЕАЭС)8. Допускается представление как МФСФ, так и КХСФ, содержание и структура которых должны соответствовать требованиям Правил надлежащей практики фармаконадзора ЕАЭС [2]. В разделе 1.10.2 РД необходимо представлять письменное подтверждение ДРУ факта наличия уполномоченного лица, ответственного за фармаконадзор на территории государств — членов ЕАЭС (рис. 1).
В ходе экспертизы РД в рамках процедуры приведения в соответствие переоценка соотношения «польза–риск» не проводится, за исключением случаев, когда заявителем указано, что в дальнейшем планируется регистрация по процедуре взаимного признания в государствах — членах ЕАЭС, в которых данный ЛП не был зарегистрирован [9]. В этой ситуации ДРУ, не имеющий на территории ЕАЭС действующей регистрации, осуществленной ранее, обязан в РД представлять МФСФ. В остальных случаях возможно представление КХСФ.
Если одновременно с процедурой приведения РД ЛП в соответствие с требованиями ЕАЭС планируется регистрация по процедуре взаимного признания в государствах — членах ЕАЭС, в которых данный ЛП не был зарегистрирован, в соответствии с п. 183 Правил регистрации и экспертизы ЕАЭС эксперт проводит переоценку соотношения «польза–риск», то к документам о СФ предъявляются те же требования, как при регистрации ЛП. Таким образом, заявителю необходимо учитывать наличие или отсутствие действующей регистрации ЛП для данного ДРУ в странах ЕАЭС.
Значимые изменения в СФ, такие как смена УЛФ или адреса осуществления деятельности по фармаконадзору, должны быть внесены как в МФСФ, представленный в РД ЛП, который является для ДРУ первым зарегистрированным для применения на фармацевтическом рынке ЕАЭС, так и в КХСФ, представленную в составе РД последующих регистрируемых ЛП. Соответствие информации в этих документах имеет важное значение, так как позволяет сделать заключение, удовлетворяет ли СФ ДРУ требованиям ЕАЭС, либо есть необходимость в проведении внеплановой инспекции СФ.
При внесении изменений в МФСФ либо КХСФ эти документы необходимо актуализировать в РД всех ЛП ДРУ. Заявителю необходимо воспользоваться классификацией изменений в РД ЛП (дополнение V к Правилам регистрации и экспертизы ЕАЭС). В документы по фармаконадзору в РД могут быть внесены: А «Административные изменения», В «Изменения безопасности, эффективности и фармаконадзора», а также неклассифицированные изменения II типа, связанные с добавлением стран признания, в которых данный ЛП не был зарегистрирован.
При внесении изменения А.1 «Изменение держателя регистрационного удостоверения» следует предоставлять МФСФ либо КХСФ, а также информацию в разделе об ответственном лице по фармаконадзору на территории ЕАЭС: как в случае изменения названия и(или) адреса ДРУ (если данная информация представлена в указанных документах) — изменение А.1.а, тип IАНУ (необходима немедленная подача заявления о внесении изменений — в течение 20 рабочих дней с даты реализации указанных изменений), так и при трансфере РД от одного ДРУ другому юридическому лицу — изменение А.1.б, тип IАНУ (табл. 1).
Таблица 1. Документы о системе фармаконадзора, представляемые держателем регистрационного удостоверения лекарственного препарата при внесении изменений, являющихся следствием другого изменения (в соответствии с Решением Совета Евразийской экономической комиссии от 03.11.2016 № 78 «О Правилах регистрации и экспертизы лекарственных средств для медицинского применения»)
Table 1. Documents on the pharmacovigilance system submitted by the marketing authorisation holder when making changes resulting from another change (Decision No. 78 of the Council of the Eurasian Economic Commission On the Rules of Marketing Authorisation and Assessment of Medicinal Products for Human Use dated 03.11.2016)
Код, тип Code, type | Заявленные изменения Declared changes | Документы для представления Required documentation |
А.1а, IАНУ* А.1а, IАIN* | Изменение названия и(или) адреса держателя регистрационного удостоверения Change in the name and/or address of the marketing authorisation holder (MAH) | 1.10.1 — 07001 мастер-файл системы фармаконадзора (07002 краткая характеристика системы фармаконадзора); 1.10.2 — 07003 уведомление 1.10.1: 07001 Pharmacovigilance System Master File (07002 Summary of the pharmacovigilance system); 1.10.2: 07003 Written confirmation by the MAH that there is a qualified person responsible for pharmacovigilance (QPPV) at their disposal in a Member State |
А.1б, IАНУ А.1b, IАIN | Трансфер регистрационного удостоверения от одного держателя регистрационного удостоверения другому юридическому лицу Transfer of marketing authorisation from the MAH to another legal entity | 1.10.1 — 07001 мастер-файл системы фармаконадзора (07002 краткая характеристика системы фармаконадзора); 1.10.2 — 07003 уведомление 1.10.1: 07001 Pharmacovigilance System Master File (07002 Summary of the pharmacovigilance system); 1.10.2: 07003 Written confirmation by the MAH that there is a QPPV at their disposal in a Member State |
Неклассифицированное изменение, тип II** Unclassifiable change II ** | Добавление стран признания, в которых лекарственный препарат не был зарегистрирован (расширение географии) Addition of a Member State concerned in which the medicinal product has not been authorised (geographic growth) | Актуализация при необходимости: 1.10.1 — 07001 мастер-файл системы фармаконадзора (07002 краткая характеристика системы фармаконадзора); 1.10.2 — 07003 уведомление Update if necessary: 1.10.1: 07001 Pharmacovigilance System Master File (07002 Summary of the pharmacovigilance system); 1.10.2: 07003 Written confirmation by the MAH that there is a QPPV at their disposal in a Member State |
Таблица составлена авторами по данным нормативного документа / The table is prepared by the authors using the regulatory document
* Незначимое изменение IAНУ типа – изменение IA типа, требующее немедленного уведомления, то есть немедленной подачи заявления о внесении изменений (в течение 20 рабочих дней с даты реализации указанных изменений).
** Значимое изменение II типа – изменение, которое, не являясь расширением регистрации, может оказать значительное влияние на качество, безопасность или эффективность зарегистрированного лекарственного препарата.
* An minor type IAIN variation is a type IA variation that requires immediate notification, that is, immediate submission of an application for the variation (within 20 working days from the date of implementation of these changes).
** A major type II variation is a variation that is not an extension of the marketing authorisation and that may have a significant impact on the quality, safety, or effectiveness of the authorised medicinal product.
Внесение изменений, связанных с документами о СФ, регламентировано требованиями подраздела B.I «Лекарственные препараты для медицинского применения» (табл. 2). Так, изменение B.I.8 «Введение или изменение резюме системы фармаконадзора лекарственного препарата для медицинского применения» (B.I.8.а, тип IАНУ) предполагает обновление КХСФ, а также обновление информации в разделе об ответственном лице на территории ЕАЭС. B.I.9 «Изменение существующей системы фармаконадзора согласно подробному описанию системы фармаконадзора (ПОСФ)» необходимо использовать для изменения МФСФ, а также для обновления информации в разделе об ответственном лице по фармаконадзору на территории ЕАЭС в следующих случаях:
Таблица 2. Документы о системе фармаконадзора, представляемые держателем регистрационного удостоверения лекарственного препарата при внесении изменений, связанных с системой фармаконадзора (в соответствии с Решением Совета Евразийской экономической комиссии от 03.11.2016 № 78 «О Правилах регистрации и экспертизы лекарственных средств для медицинского применения»)
Table 2. Documents on the pharmacovigilance system submitted by the marketing authorisation holder when making changes associated with the pharmacovigilance system (Decision No. 78 of the Council of the Eurasian Economic Commission On the Rules of Marketing Authorisation and Assessment of Medicinal Products for Human Use dated 03.11.2016)
Код, тип Code, type | Заявленные изменения Declared changes | Документы для представления Required documentation |
В.I.8а, IАНУ* С.I.8а, IАIN* | Введение резюме системы фармаконадзора, изменений квалифицированного лица по фармаконадзору (включая контактную информацию) и (или) изменение месторасположения мастер-файла системы фармаконадзора Introduction of a summary of the pharmacovigilance system, changes related to the qualified person responsible for pharmacovigilance (QPPV) (including contact details), and/or changes in the Pharmacovigilance System Master File (PSMF) location | 1.10.1 – 07002 краткая характеристика системы фармаконадзора; 1.10.2 – 07003 уведомление 1.10.1: 07002 Summary of the pharmacovigilance system; 1.10.2: 07003 Written confirmation by the MAH that there is a QPPV at their disposal in a Member State |
В.I.9а, IАНУ С.I.9а, IАIN | Изменение квалифицированного лица по фармаконадзору, и(или) контактной информации, и(или) процедуры резервирования Change related to the QPPV and/or QPPV contact details and/or back-up procedure | 1.10.1 — 07001 мастер-файл системы фармаконадзора; 1.10.2 — 07003 уведомление 1.10.1: 07001 PSMF; 1.10.2: 07003 Written confirmation by the MAH that there is a QPPV at their disposal in a Member State |
В.I.9б, IАНУ С.I.9b, IАIN | Изменение баз данных и(или) основных контрактных соглашений Change(s) in the safety database and/or major contractual arrangements for the fulfilment of pharmacovigilance obligations, and/or change of the site undergoing pharmacovigilance activities | 1.10.1 — 07001 мастер-файл системы фармаконадзора 1.10.1: 07001 PSMF |
В.I.9в, IА** С.I.9с, IА** | Иные изменения мастер-файла системы фармаконадзора Other change(s) to the PSMF | 1.10.1 — 07001 мастер-файл системы фармаконадзора 1.10.1: 07001 PSMF |
В.I.9г, IАНУ С.I.9d, IАIN | Внесение изменений по результатам экспертизы другого лекарственного препарата этого держателя регистрационного удостоверения Change(s) following an assessment in relation to another medicinal product of the MAH | 1.10.1 — 07001 мастер-файл системы фармаконадзора 1.10.1: 07001 PSMF |
Таблица составлена авторами по данным нормативного документа / The table is prepared by the authors using the regulatory document
* Незначимое изменение IAНУ типа — изменение IA типа, требующее немедленного уведомления, то есть немедленной подачи заявления о внесении изменений (в течение 20 рабочих дней с даты реализации указанных изменений).
** Незначимое изменение IA типа — изменение, которое оказывает минимальное влияние на качество, безопасность и эффективность зарегистрированного лекарственного препарата или не оказывает его.
* An minor type IAIN variation is a type IA variation that requires immediate notification, that is, immediate submission of an application for the variation (within 20 working days from the date of implementation of these changes).
** An minor type IA variation is a variation that has only a minimal impact or no impact at all on the quality, safety, or effectiveness of the authorised medicinal product.
Порядок представления документов о СФ ДРУ зафиксирован в нормативных правовых актах в сфере обращения лекарственных средств в рамках ЕАЭС. Однако заявители нередко представляют в составе РД иные документы. В разделе 1.10.1 вместо МФСФ ДРУ заявители размещают МФСФ производителя (отличного от ДРУ), МФСФ заявителя (отличного от ДРУ), МФСФ аутсорсинговой компании, в которых не указана информация о СФ ДРУ. Документ о СФ может быть составлен не в соответствии с требованиями надлежащей практики фармаконадзора, иметь отсылки к недействующим нормативным правовым актам либо к законодательным актам государств, не входящих в ЕАЭС. Нарушением является также размещение в разделе 1.10 РД различных деклараций, пояснений о СФ, стандартов предприятий, текстов стандартных операционных процедур компаний и других документов, не предназначенных для описания СФ ДРУ. Распространенной ошибкой является указание неверного кода документа: для КХСФ указывают код 07001 (должен быть 07002), а для МСФС — код 07002 (должен быть 07001), что нарушает порядок расположения этих документов в составе РД.
При процедуре приведения РД ЛП в соответствие с требованиями ЕАЭС наиболее распространенной ошибкой в документах о СФ являются разночтения в информации относительно УЛФ ЕАЭС. Например, представлены МФСФ и/ или КХСФ, содержащие данные об одном УЛФ, а в документе об ответственном за фармаконадзор на территории государств — членов ЕАЭС указаны данные иного УЛФ ЕАЭС. Возможны иные вариации, когда МФСФ и представленная КХСФ одного ДРУ содержат данные о разных УЛФ ЕАЭС.
Зачастую представленные документы о СФ принадлежат аутсорсинговой организации, которой были переданы полномочия по осуществлению фармаконадзора компанией ДРУ, но при этом отсутствуют информация о самом ДРУ и доказательства полномочий, позволяющие представлять интересы ДРУ как компании, отвечающей за фармаконадзор на территории государств — членов ЕАЭС. В данном случае необходимо помнить, что в соответствии с Приложением № 1 Решения Совета ЕЭК № 78 от 03.11.2016 и Решением Совета ЕЭК от 03.11.2016 № 87 МФСФ должен описывать СФ, разработанную и внедренную в работу компании ДРУ [10].
В единичных случаях в РД полностью отсутствуют документы о СФ и письменное подтверждение ДРУ факта наличия уполномоченного лица, ответственного за фармаконадзор, что также является нарушением требований законодательства ЕАЭС.
Возможной причиной описанных ошибок является несвоевременная актуализация данных в документах о СФ либо не организована эффективная система взаимодействия между сотрудниками отдела по фармаконадзору и сотрудниками отдела регистрации, занимающимися комплектацией документов РД для подачи в экспертную организацию. В ситуации, когда документ находится в постоянном неконтролируемом доступе, существует необходимость пересмотреть процедуру предоставления информации о документах СФ сотрудникам иных подразделений, в частности отдела регистрации, при возможности — ограничить доступ, провести соответствующее обучение.
Ошибки в случае внесения изменений в РД могут касаться как самого документа о СФ (в том числе его непредставление), так и кодировки изменения. Так, ДРУ может заявить об изменении документов о СФ, но не загрузить новый документ в РД, или же не представить документ, изменение которого является следствием заявленного изменения (название ДРУ, трансфер ДРУ). Достаточно часто встречается несоответствие заявленного кода самому документу (например, с кодом В.I.8а представляют МФСФ), а также неверное указание типа (например, IВ неклассифицируемое). Отдельно следует отметить случай, когда документы были загружены, но информация об этом отсутствовала в заявлении о внесении изменений.
Подобные ошибки свидетельствуют о том, что заявитель не в полной мере изучил Правила регистрации и экспертизы ЕАЭС. При выявлении несоответствий порядка представления, содержания, кодирования документов о СФ регуляторными органами формируется запрос к ДРУ о предоставлении недостающей информации и устранении недочетов. Неустранение ДРУ замечаний или непредставление в течение отведенного времени ответов на вопросы, возникшие во время проведения экспертизы документов о СФ, является основаниями для отказа в подтверждении регистрации (перерегистрации) ЛП.
При подготовке документов о СФ в РД необходимо соблюдать требования Правил надлежащей практики фармаконадзора ЕАЭС (Решение Совета ЕЭК от 03.11.2016 № 87) и Правил регистрации и экспертизы лекарственных средств для медицинского применения (Решение Совета ЕЭК от 03.11.2016 № 78). Cтруктура МФСФ и КХСФ должна соответствовать требованиям Правил надлежащей практики фармаконадзора ЕАЭС, а содержание — отражать реальную деятельность по фармаконадзору ДРУ. Для того чтобы избежать постоянных обновлений РД, связанных с появлением новой версии МФСФ, заявителю достаточно представить данный документ в первом РД ЛП от данного ДРУ, а в дальнейшем представлять КХСФ как для процедуры регистрации ЛП, так и для процедуры приведения РД ЛП в соответствие с требованиями ЕАЭС.
При внесении изменений в МФСФ и КХСФ в срок, указанный в нормативном документе, следует уведомлять регуляторные органы. Изменения документов о СФ в основном относятся к типу IАНУ — незначимые изменения, требующие немедленного уведомления в течение 20 рабочих дней с даты реализации. Обновление данных по фармаконадзору требуется не только в случае непосредственного внесения изменений в документы по фармаконадзору, но и в случае, когда в заявлении указаны изменения, которые требуют пересмотра информации в разделах РД по фармаконадзору.
Технические ошибки и представление неактуальной информации в разделах РД ДРУ можно минимизировать, организовав взаимодействие между сотрудниками отдела по фармаконадзору и сотрудниками отдела регистрации, а также обеспечив контролируемый доступ к документам СФ.
Соблюдение порядка представления и соответствия содержания МФСФ и КХСФ требованиям законодательных актов ЕАЭС позволит заявителю осуществлять эффективный фармаконадзор, а также быть готовым к проведению инспекции СФ со стороны регуляторных органов.
Вклад авторов. Все авторы подтверждают соответствие своего авторства критериям ICMJE. Наибольший вклад распределен следующим образом: Н.Ю. Вельц — идея, планирование, написание и редактирование текста рукописи, утверждение окончательной версии рукописи для публикации; Е.О. Журавлева и Г.В. Кутехова — написание отдельных разделов, редактирование текста рукописи, формулировка выводов; Н.В. Терешкина — обобщение результатов, формулировка выводов.
Authors’ contributions. All the authors confirm that they meet the ICMJE criteria for authorship. The most significant contributions were as follows. Nataliya Yu. Velts conceived the idea, planned the study, drafted and edited the manuscript, and approved the final version for publication. Evgeniya O. Zhuravleva and Galina V. Kutekhova drafted individual sections of the manuscript, edited the manuscript, and formulated the conclusions. Nataliya V. Tereshkina summarised the results and formulated the conclusions.
1. Решение Совета Евразийской экономической комиссии от 03.11.2016 № 87 «Об утверждении Правил надлежащей практики фармаконадзора Евразийского экономического союза».
2. Там же.
3. Решение Совета Евразийской экономической комиссии от 19.05.2022 № 81 «О внесении изменений в Правила надлежащей практики фармаконадзора Евразийского экономического союза».
Решение Совета Евразийской экономической комиссии от 03.11.2016 № 87 «Об утверждении Правил надлежащей практики фармаконадзора Евразийского экономического союза».
4. Решение Совета Евразийской экономической комиссии от 19.05.2022 № 81 «О внесении изменений в Правила надлежащей практики фармаконадзора Евразийского экономического союза».
5. Решение Совета Евразийской экономической комиссии от 03.11.2016 № 87 «Об утверждении Правил надлежащей практики фармаконадзора Евразийского экономического союза».
Решение Совета Евразийской экономической комиссии от 03.11.2016 № 78 «О Правилах регистрации и экспертизы лекарственных средств для медицинского применения».
6. Решение Коллегии Евразийской экономической комиссии от 17.09.2019 № 159 «О классификаторе видов документов регистрационного досье лекарственного препарата и справочнике структурных элементов регистрационного досье лекарственного препарата».
7. Решение Коллегии Евразийской экономической комиссии от 17.09.2019 № 159 «О классификаторе видов документов регистрационного досье лекарственного препарата и справочнике структурных элементов регистрационного досье лекарственного препарата».
Решение Совета Евразийской экономической комиссии от 03.11.2016 № 78 «О Правилах регистрации и экспертизы лекарственных средств для медицинского применения».
8. Решение Совета Евразийской экономической комиссии от 20.10.2023 № 114 «О внесении изменений в Правила регистрации и экспертизы лекарственных средств для медицинского применения».
1. Снегирева ИИ, Журавлева ЕО, Вельц НЮ. Экспертная оценка документов о системе фармаконадзора в составе регистрационного досье. Безопасность и риск фармакотерапии. 2020;8(4):191–7. https://doi.org/10.30895/2312-7821-2020-8-4-191-197
2. Вельц НЮ, Журавлева ЕО, Кутехова ГВ, Терешкина НВ. Мастер-файл системы фармаконадзора: обзор изменений в Правилах надлежащей практики фармаконадзора ЕАЭС. Безопасность и риск фармакотерапии. 2023;11(1):22–9. https://doi.org/10.30895/2312-7821-2023-11-1-22-29
3. Таубэ АА, Евко ИЮ, Синотова СВ, Крашенинников АЕ, Журавлева МВ, Романов БК, Аляутдин РН. Российский фармаконадзор: пути повышения эффективности. Вестник Российской военномедицинской академии. 2022;24(1):81–90. https://doi.org/10.17816/brmma89665
4. Таубэ АА, Романов БК. Аудиты и инспекции систем фармаконадзора в России. Качественная клиническая практика. 2023;(1):4–14. https://doi.org/10.37489/2588-0519-2023-1-4-14
5. Patel P, Badjatya JK, Hinge M. Comparative study of regulatory requirements of drug product in emerging market. Int J Drug Reg Affairs. 2019;7(3):48–2. https://doi.org/10.22270/ijdra.v7i3.350
6. Potts J, Genov G, Segec A, Raine J, Straus S, Arlett P. Improving the safety of medicines in the European Union: from signals to action. Clin Pharmacol Ther. 2020;107(3):521–9. https://doi.org/10.1002/cpt.1678
7. Журавлева ЕО, Вельц НЮ, Кутехова ГВ. Анализ несоответствий требованиям законодательства ЕАЭС в документах по фармаконадзору в составе регистрационного досье. Безопасность и риск фармакотерапии. 2021;9(4):185–90. https://doi.org/10.30895/2312-7821-2021-9-4-185-190
8. Рычихина ЕМ, Ткаченко ОГ, Косенко ВВ. Рекомендации для специалистов по регистрации лекарственных препаратов в целях оптимизации работ по процедурам ЕАЭС. Ведомости Научного центра экспертизы средств медицинского применения. Регуляторные исследования и экспертиза лекарственных средств. 2023;13(2–1):345–60. https://doi.org/10.30895/1991-2919-2023-544
9. Таубэ АА, Левашова АЮ. Приведение регистрационного досье на лекарственный препарат в соответствие с требованиями Евразийского экономического союза. Вопросы обеспечения качества лекарственных средств. 2020;2(28):40–7. https://doi.org/10.34907/JPQAI.2020.22.25.006
10. Ситникова ЕА, Марданлы СГ, Рогожникова ЕП. Система фармаконадзора на реальном предприятии. Разработка и регистрация лекарственных средств. 2018;(2):170–2.
Вельц Наталья Юрьевна, канд. биол. наук, доцент
Петровский б-р, д. 8, стр. 2, Москва, 127051
Журавлева Евгения Олеговна
Петровский б-р, д. 8, стр. 2, Москва, 127051
Кутехова Галина Викторовна
Петровский б-р, д. 8, стр. 2, Москва, 127051
Терешкина Наталия Васильевна, канд. мед. наук
Петровский б-р, д. 8, стр. 2, Москва, 127051
Вельц Н.Ю., Журавлева Е.О., Кутехова Г.В., Терешкина Н.В. Представление документов о системе фармаконадзора в составе регистрационного досье в рамках процедур ЕАЭС: анализ требований и типичных ошибок. Безопасность и риск фармакотерапии. 2024;12(3):331-340. https://doi.org/10.30895/2312-7821-2024-12-3-331-340
Velts N.Yu., Zhuravleva E.O., Kutekhova G.V., Tereshkina N.V. Submission of Documents on the Pharmacovigilance System as Part of the Registration Dossier within the Framework of the EAEU Procedures: Analysis of Requirements and Typical Errors. Safety and Risk of Pharmacotherapy. 2024;12(3):331-340. (In Russ.) https://doi.org/10.30895/2312-7821-2024-12-3-331-340
Издатель: ФГБУ «НЦЭСМП» Минздрава России
Обработка персональных данных
































