


ГЛАВНАЯ ТЕМА: ИННОВАЦИИ И ВЫЗОВЫ В ФАРМАКОТЕРАПИИ: ОТ РАЗРАБОТКИ НОВЫХ ПРЕПАРАТОВ ДО ОБЕСПЕЧЕНИЯ БЕЗОПАСНОСТИ В КЛИНИЧЕСКОЙ ПРАКТИКЕ
ВВЕДЕНИЕ. Болезнь Альцгеймера, распространение которой коррелирует с увеличением продолжительности жизни населения, является одной из главных причин тяжелых когнитивных расстройств и деменции. В 2021–2024 гг. Управлением по контролю качества пищевых продуктов и лекарственных средств (FDA) были зарегистрированы болезнь-модифицирующие препараты на основе антиамилоидных (анти-Аβ) моноклональных антител (МкАТ): адуканумаб (ускоренная регистрация), леканемаб и донанемаб. Изучение эффективности и безопасности этих препаратов продолжается.
ЦЕЛЬ. Оценка перспектив и ограничений антиамилоидной терапии болезни Альцгеймера болезнь-модифицирующими препаратами в контексте современных представлений о патогенетических механизмах этого заболевания.
ОБСУЖДЕНИЕ. По современным представлениям в патогенезе болезни Альцгеймера основную роль отводят отложению в головном мозге амилоидных бляшек и нейрофибриллярных клубков из патологического гиперфосфорилированного тау-белка, что сопровождается нейродегенеративными изменениями. Патогенез заболевания продолжает изучаться. Механизм действия одобренных FDA для лечения болезни Альцгеймера анти-Аβ МкАТ адуканумаб, леканемаб и донанемаб — активация микроглии с фагоцитозом амилоида и его деградацией; различие между ними заключается в аффинности к разным формам амилоида. Результаты клинических исследований (средняя продолжительность 1,5 года) показали, что все анти-Аβ МкАТ достоверно и значимо уменьшали амилоидную нагрузку в головном мозге (вплоть до полного ее исчезновения при применении донанемаба), а также замедляли когнитивные нарушения у пациентов на ранней стадии болезни Альцгеймера. Замедление когнитивных нарушений было достоверным, но клинически малозначимым. Основные осложнения терапии МкАТ — амилоид-ассоциированные аномалии визуализации (ARIA), определявшиеся у 20–30% пациентов, являются результатом удаления амилоида, чаще возникают в начале лечения и у носителей аллеля APOE ε4. В большинстве случаев ARIA бессимптомны и регрессируют со временем.
ВЫВОДЫ. Анти-Аβ МкАТ достоверно показали эффективность в уменьшении амилоидной нагрузки головного мозга и замедлении прогрессирования когнитивных расстройств при болезни Альцгеймера. Однако малая клиническая эффективность, инвазивность диагностики, высокая стоимость диагностики и терапии, дополнительные расходы на мониторинг нежелательных явлений в настоящее время ограничивают широкое применение препаратов этой группы.
ВВЕДЕНИЕ. В связи с прекращением с 1980 г. вакцинации против оспы после успешного проведения глобальной программы Всемирной организации здравоохранения по ее ликвидации в настоящее время более половины населения Земли лишено иммунитета в отношении патогенных для человека ортопоксвирусов. Вспышки оспы обезьян в нескольких странах с 2022 г. свидетельствуют об актуальности разработки новых химических препаратов, обладающих антиортопоксвирусной активностью.
ЦЕЛЬ. Оценить безопасность и переносимость российского противооспенного препарата НИОХ-14 при пероральном применении в клиническом исследовании I фазы.
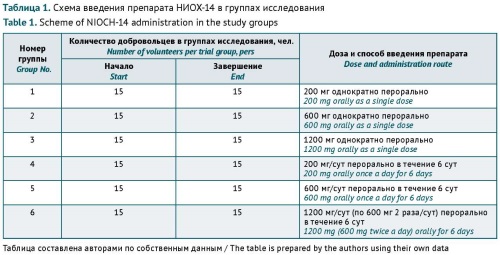
МАТЕРИАЛЫ И МЕТОДЫ. Исследование проведено на 90 здоровых добровольцах на базе ФГБУЗ «Медико-санитарная часть № 163 Федерального медико-биологического агентства» по протоколу «Открытое простое рандомизированное исследование безопасности, переносимости, фармакокинетики препарата НИОХ-14 на добровольцах в возрасте 18–50 лет в параллельных группах» № NIOCH-01/20. В работе использовали стандартный комплекс клинических и лабораторно-инструментальных методов. В исследовании принимали участие 6 групп по 15 добровольцев: 1, 2 и 3 группы получали однократно перорально 200, 600 и 1200 мг НИОХ-14 соответственно; 4, 5 и 6 группы получали ежедневно перорально по 200, 600 и 1200 мг НИОХ-14 соответственно в течение 6 сут.
РЕЗУЛЬТАТЫ. При однократном и многократном применении препарата в дозах 200, 600 и 1200 мг не выявлено изменений в крови добровольцев при анализе следующих показателей в динамике: эритроциты, лейкоциты, тромбоциты, лейкоцитарная формула, гемоглобин, скорость оседания эритроцитов, аланиновая и аспарагиновая трансаминазы, щелочная фосфатаза, общий белок, общий билирубин, глюкоза, креатинин, мочевина, С-реактивный белок, протромбиновый индекс и тимоловая проба. Не установлено изменений показателей общего анализа мочи в течение всего периода наблюдения. Не выявлено случаев возникновения нежелательных явлений при однократном применении НИОХ-14 в исследуемых дозах и при многократном применении в дозах 200 и 600 мг. При многократном приеме препарата НИОХ-14 в дозе 1200 мг у одного добровольца на 2-е и 5-е сутки выявлена кратковременная болезненность в эпигастральной области.
ВЫВОДЫ. При анализе данных физикального обследования, клинического и биохимического анализов крови и мочи здоровых добровольцев патологических изменений не выявлено. Препарат НИОХ-14 в форме твердых желатиновых капсул, содержащих 200 мг НИОХ-14, обладает высоким профилем безопасности и переносимости.

ВВЕДЕНИЕ. Использование релевантных видов экспериментальных животных для проведения доклинических исследований безопасности при разработке нового лекарственного средства позволяет получить значимую информацию для оценки пользы и риска применения препарата у человека. Выбор подходящих видов включает рассмотрение научных, этических и практических аспектов и требует тщательного обоснования. В нормативных документах Евразийского экономического союза (ЕАЭС) содержатся указания на необходимость проведения доклинических исследований безопасности лекарственных средств с использованием релевантных животных, однако имеющиеся рекомендации являются недостаточными. Актуально определение значимых критериев выбора видов экспериментальных животных на основании анализа международных нормативных документов по доклиническим исследованиям и рекомендаций научного сообщества.
ЦЕЛЬ. Анализ нормативной правовой и научно-методической базы, определение ключевых факторов и критериев для обоснования выбора релевантных видов экспериментальных животных при проведении доклинических исследований безопасности лекарственных средств.
ОБСУЖДЕНИЕ. Проанализированы руководства ЕАЭС, Международного совета по гармонизации технических требований к фармацевтическим препаратам для использования человеком (ICH), Европейского агентства по лекарственным средствам (EMA), регламентирующие доклинические исследования, и научные публикации по выбору экспериментальных животных. Установлено, что рекомендации по выбору релевантных животных наиболее полно отражены в Правилах проведения исследований биологических лекарственных препаратов ЕАЭС и ICH S6(R1), руководствах ICH S5(R3) по изучению репродуктивной токсичности, ICH S11 по разработке педиатрических препаратов и EMA по стратегиям идентификации и минимизации риска при первом применении препарата у человека. Вопросы выбора подходящих животных являются предметом оживленной научной дискуссии. Спонсоры исследований отмечают, что большинство запросов регуляторного органа, связанных с релевантностью, касаются предоставления дополнительной информации о фармакологической значимости животных, обоснования использования только одного вида, а также рекомендаций по проведению дополнительных исследований на других видах животных. Многие исследовательские коллективы руководствуются в своей работе внутренними документами, в которых описаны этапы и критерии выбора релевантных животных. Научным сообществом предложено более сорока различных показателей, оценка которых в опытах in vitro, in vivo и in silico позволяет обосновать релевантность экспериментальных животных для доклинических исследований безопасности лекарственных средств.
ВЫВОДЫ. Выбор релевантных тест-систем и моделей в доклинических исследованиях безопасности лекарственных средств является самостоятельной научной задачей. Для обоснования релевантности экспериментальных животных, обеспечения трансляционности полученных результатов и соблюдения этических принципов наибольшую ценность представляют критерии, разработанные на основе системного подхода, который базируется на анализе комплекса фармакодинамических, фармакокинетических и токсикологических параметров in vitro и in vivo.
ВВЕДЕНИЕ. В связи с обнаружением новых рисков, обусловленных применением фторхинолонов и распространением антибиотикорезистентности, большое значение для обеспечения рациональной антибиотикотерапии приобретают выявление и анализ медицинских ошибок при назначении препаратов данной группы. Одним из наиболее часто используемых в реальной клинической практике фторхинолонов является левофлоксацин.
ЦЕЛЬ. Изучить структуру медицинских ошибок при применении препаратов группы фторхинолонов на примере левофлоксацина на основе анализа данных российской базы спонтанных сообщений.
МАТЕРИАЛЫ И МЕТОДЫ. Проведен ретроспективный анализ спонтанных сообщений, поступивших в российскую базу данных, о нежелательных реакциях в период с 01.04.2019 по 28.02.2023. Критерии включения: спонтанные сообщения, по информации которых нежелательные реакции произошли на территории Российской Федерации и в качестве подозреваемого препарата указан левофлоксацин. Для выявления медицинских ошибок использовали информацию утвержденных инструкций по медицинскому применению левофлоксацина, официальных клинических рекомендаций и Программы СКАТ (Стратегия контроля антимикробной терапии).
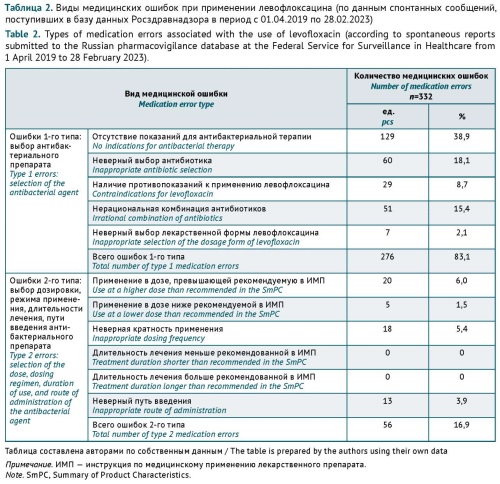
РЕЗУЛЬТАТЫ. В анализ было включено 950 спонтанных сообщений. Факты медицинских ошибок при назначении левофлоксацина были выявлены в 307 (32,3%) спонтанных сообщениях, общее количество ошибок — 332. Основными видами медицинских ошибок при выборе препарата являлись: назначение левофлоксацина при отсутствии показаний для любой антибактериальной терапии (38,9% от всех медицинских ошибок, n=129, при этом в 76,0% случаев левофлоксацин применяли при вирусных инфекциях), неверный выбор левофлоксацина в качестве препарата первой линии (18,1%, n=60), нерациональное, в основном избыточное, назначение комбинации левофлоксацина с другими антибактериальными препаратами (15,4%, n=51). Реже медицинские ошибки заключались в нарушениях режима дозирования (13,0%, n=43), назначении левофлоксацина при наличии противопоказаний (8,7%, n=29), неверном выборе пути введения (3,9%, n=13) и лекарственной формы (2,1%, n=7).
ВЫВОДЫ. Практика назначения антибиотиков, в частности левофлоксацина, при вирусных инфекциях продолжается, несмотря на убедительные доказательства отсутствия эффекта. Эффективное и безопасное использование антибиотиков обеспечивает только их назначение по утвержденным показаниям с соблюдением рекомендуемой дозировки и способа применения. Необходима разработка мер по ограничению избыточного применения антибиотиков, которое может иметь неблагоприятные последствия не только для конкретного пациента, но и для общественного здоровья в целом в связи с повышением риска развития микробной резистентности.
ВВЕДЕНИЕ. Лекарственно-индуцированное поражение печени (ЛИПП) является распространенным осложнением при применении лекарственных препаратов и биологически активных добавок к пище, которое может приводить к жизнеугрожающим последствиям. Выявление признаков поражения печени на раннем этапе может быть затруднено при наличии полипрагмазии, коморбидности, фоновых заболеваний печени у пациента, а также в связи с неправильной трактовкой неспецифической клинической картины нарушений. Обобщение информации о возможностях диагностики и прогностической оценки ЛИПП для врачей амбулаторного звена является актуальным.
ЦЕЛЬ. Обобщение и систематизация информации о факторах риска развития, методах ранней диагностики и прогностической оценки для подготовки рекомендаций по выявлению ЛИПП в амбулаторных условиях.
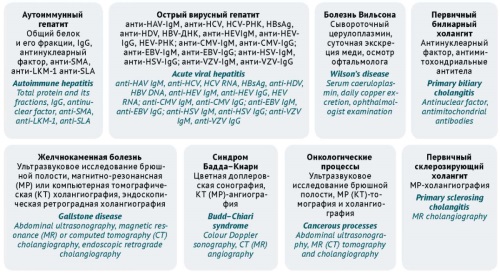
ОБСУЖДЕНИЕ. Основными факторами риска, способствующими возникновению ЛИПП, являются применение потенциально гепатотоксичных лекарственных препаратов, высокая доза препарата, полипрагмазия, заболевания печени в анамнезе, генетическая предрасположенность. ЛИПП потенциально способно вызвать применение любого лекарственного препарата, но особое значение имеет осведомленность врачей амбулаторного звена о препаратах, наиболее часто индуцирующих данное состояние. При первичном скрининге повреждения печени и определения степени его тяжести необходимы тщательный сбор лекарственного анамнеза и проведение стандартных лабораторных тестов (активность аланиновой трансаминазы, аспарагиновой трансаминазы, щелочной фосфатазы, уровень общего билирубина). В амбулаторных условиях актуальным является использование шкал для выявления причинно-следственной связи между применением лекарственного препарата и развитием нарушений (CIOMS/RUCAM, M&V, DDW—J, Наранжо), а также инструментов для прогностической оценки и оценки степени тяжести ЛИПП (закон Хая). Следует учитывать, что шкала CIOMS/RUCAM подходит при подозрении на гепатоцеллюлярное или холестатическое ЛИПП, шкала M&V адаптирована также для смешанного варианта ЛИПП и позволяет учитывать внепеченочные проявления.
ВЫВОДЫ. Рекомендованный подход, включающий, помимо сбора анамнеза, осмотра, лабораторной и инструментальной диагностики, рутинное использование шкал для выявления причинно-следственной связи между приемом препарата и развитием ЛИПП и прогностической оценки состояния пациента, будет способствовать выявлению поражения печени врачами амбулаторного звена на раннем этапе и быстрому принятию решений о дальнейшем лечении.
ВВЕДЕНИЕ. Антипсихотик-индуцированный паркинсонизм (АИП) — неврологическая нежелательная реакция со стороны экстрапирамидной системы, возникающая на фоне приема антипсихотиков (АП), которая относится к несерьезным нежелательным реакциям. Однако при развитии АИП у пациентов с расстройствами шизофренического спектра значительно снижается качество жизни, поэтому актуальной задачей является ранняя диагностика и своевременная коррекция АИП.
ЦЕЛЬ. Разработать шкалу оценки риска и персонализированный алгоритм диагностики АИП как наиболее распространенной и клинически значимой неврологической нежелательной реакции у пациентов с расстройствами шизофренического спектра.
МАТЕРИАЛЫ И МЕТОДЫ. Проведен анализ модифицируемых и немодифицируемых факторов риска развития АИП, шкал и опросников, которые используются для диагностики АИП, а также методов лабораторной диагностики АИП на основе информации из полнотекстовых публикаций на русском и английском языках, размещенных в базах данных eLIBRARY.RU, PubMed, Springer, ClinicalKey, Google Scholar. Предварительно выполнена сравнительная оценка эффективности использования валидных шкал риска развития АИП: шкала Симпсона–Ангуса (Simpson–Angus Scale, SAS), шкала оценки экстрапирамидных симптомов (Extrapyramidal Symptom Rating Scale, ESRS), унифицируемая шкала оценки болезни Паркинсона (Unified Parkinson’s Disease Rating Scale, UPDRS), шкала Хэн и Яра (H&Y Scale), рейтинговая шкала Д.Д. Вебстера (Webster Scale), рейтинговая шкала оценки паркинсонизма R.H. Mindham (Mindham Scale). Учтены такие характеристики, как продолжительность тестирования, степень достоверности оценки клинических проявлений АИП, возможность оценки факторов риска (предикторов) АИП, возможность оценки скорости развития АИП. Полученные результаты легли в основу разработки авторского рискометра АИП и алгоритма диагностики.
РЕЗУЛЬТАТЫ. Разработана авторская шкала диагностики и прогнозирования развития АИП, позволяющая оценить риск развития АИП. Для пациентов, имеющих высокий и средний риск развития АИП, определены направления персонализированной тактики ведения пациента. Представлен алгоритм диагностики АИП у пациентов с расстройствами шизофренического спектра в двух вариациях: с использованием прореактивного и реактивного фармакогенетического тестирования. Показано, что прореактивное фармакогенетическое тестирование позволяет определить риск развития АИП у пациента до применения основной терапии.
ВЫВОДЫ. Использование разработанной шкалы оценки риска и алгоритмов диагностики АИП может быть актуальным для практикующих неврологов, психиатров и клинических фармакологов. Разработка и внедрение в реальную клиническую практику новых инструментов для оценки риска, профилактики и диагностики АИП как наиболее распространенной неврологической нежелательной реакции при применении антипсихотических препаратов может обеспечить повышение качества лечебно-профилактической помощи пациентам с рассматриваемыми психическими расстройствами.
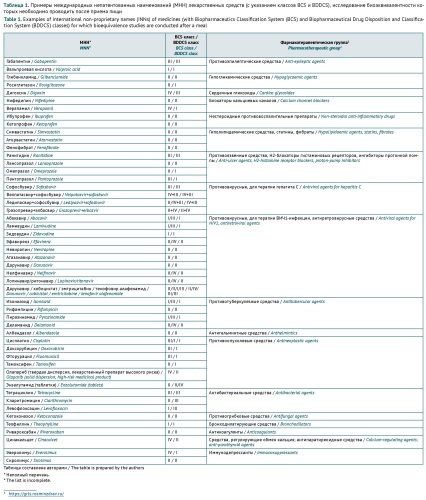
ВВЕДЕНИЕ. Актуальность изучения влияния пищи на биодоступность лекарственных препаратов (ЛП) обусловлена необходимостью правильного выбора условий применения ЛП в ходе исследований биодоступности и биоэквивалентности воспроизведенных ЛП, а также обоснования режима приема оригинальных препаратов для различных групп пациентов в клинических исследованиях. Однако в настоящее время отсутствуют единые гармонизированные требования, регулирующие изучение влияния пищи на биодоступность.
ЦЕЛЬ. Анализ нормативных национальных и международных требований, регламентирующих проведение клинических исследований биоэквивалентности для оценки условий изучения влияния пищи в исследованиях биоэквивалентности. Выявление общих требований и отличий в практике разных стран для выбора оптимальных условий проведения исследований биоэквивалентности лекарственных препаратов.
ОБСУЖДЕНИЕ. При анализе влияния пищи на биодоступность ЛП следует ориентироваться на классификации биофармацевтической классификационной системы (BCS) и биофармацевтической классификационной системы ЛП по их растворимости и метаболизму (BDDCS), определяющие растворимость, проницаемость и метаболизм ЛП. Анализ документов международных организаций (Всемирная организация здравоохранения (ВОЗ), Международный совет по гармонизации технических требований к лекарственным средствам для медицинского применения (ICH)) и регуляторных органов государств — членов ЕАЭС, стран Европы (Европейское агентство по лекарственным средствам (EMA), США (Управление по контролю за качеством продуктов питания и лекарственных средств (FDA)) и некоторых других показал различия в условиях изучения влияния пищи на биодоступность ЛП. Общим является требование проведения исследования биоэквивалентности в стандартизированных условиях. Различия заключаются в необходимом объеме исследований после приема пищи, в рекомендованном составе пищи: калорийности, содержании жиров, белков и углеводов, особенностей местных диетических предпочтений, а также в подходах к изучению влияния пищи на биодоступность ЛП высокого риска, ЛП с линейной или нелинейной фармакокинетикой и лекарственных форм с модифицированным высвобождением.
ВЫВОДЫ. Выявленные особенности национальных и международных требований к проведению клинических исследований биоэквивалентности ЛП подчеркивают целесообразность гармонизации требований нормативных документов. Это будет способствовать обеспечению безопасного и эффективного применения ЛП, а также формированию единых подходов к интерпретации результатов влияния пищи на биодоступность и выводу ЛП на международный фармацевтический рынок.
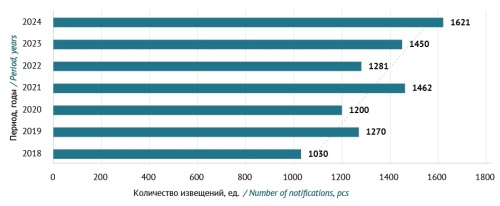
ВВЕДЕНИЕ. В пострегистрационном периоде необходимо проводить мониторинг эффективности и безопасности лекарственных препаратов для выявления, оценки и предотвращения нежелательных последствий их применения. Результативность фармаконадзорных мероприятий во многом зависит от соблюдения сотрудниками медицинских организаций правил и сроков передачи информации в контрольно-надзорные органы.
ЦЕЛЬ. Систематизация и выделение ключевых аспектов для выявления направлений деятельности по внедрению, мониторингу и совершенствованию системы фармаконадзора в медицинской организации. Анализ опыта организации фармаконадзора в медицинских организациях на примере г. Москвы за период 2018–2024 гг.
ОБСУЖДЕНИЕ. Опыт создания международной базы данных о нежелательных реакциях VigiBase, а также европейской базы данных EudraVigilance и единой информационной базы данных Евразийского экономического союза подчеркивает значимость системы фармаконадзора и актуальность ее совершенствования. Основными инструментами фармаконадзора являются спонтанные сообщения, сообщения о подозреваемых нежелательных реакциях, полученные по запросу, и активный надзор. Росздравнадзором разработаны четкие критерии работы медицинской организации по фармаконадзору: порядок сбора, регистрации информации о нежелательных реакциях и передачи сведений в Росздравнадзор, назначение ответственного за фармаконадзор, получение доступа в подсистему «Фармаконадзор» автоматизированной информационной системы Росздравнадзора (АИС) Росздравнадзора должны быть определены локальными актами. Подробно рассмотрены инструменты для создания эффективной системы фармаконадзора в медицинских организациях. На основе опыта субъекта Российской Федерации выделены и представлены основные направления организации фармаконадзора в медицинских организациях.
ВЫВОДЫ. Для повышения эффективности функционирования системы фармаконадзора в медицинской организации необходимо обеспечить качество, целостность и полноту представляемой информации при направлении спонтанных сообщений о нежелательных реакциях. Наряду с использованием спонтанного репортирования следует внедрять методы активного надзора, в том числе метод глобальных триггеров. Существующая система фармаконадзора медицинских организаций г. Москвы демонстрирует устойчивый рост репортирования в Росздравнадзор о случаях развития нежелательных реакций.
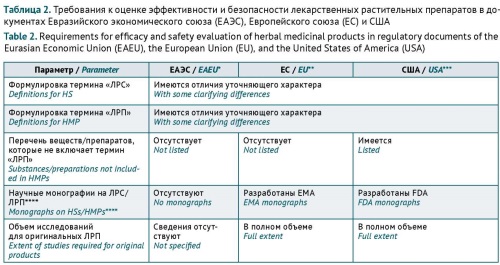
ВВЕДЕНИЕ. Лекарственные растительные препараты (ЛРП) широко применяются в современной медицинской практике. Требования к объему исследований по эффективности и безопасности ЛРП в различных странах имеют особенности, что отражено в документах, регулирующих процедуру регистрации. В Евразийском экономическом союзе (ЕАЭС) в настоящее время отсутствует руководство, регламентирующее объем доклинических и клинических исследований для ЛРП.
ЦЕЛЬ. Оценка возможности использования международных методических подходов для подготовки руководства ЕАЭС по доклиническому и клиническому изучению эффективности и безопасности ЛРП.
ОБСУЖДЕНИЕ. Особенности регистрации ЛРП связаны с содержанием в их составе сложных комплексов биологически активных веществ. Результаты анализа действующих в Европейском союзе, США и ЕАЭС подходов к оценке эффективности и безопасности препаратов растительного происхождения показали, что требования в определенной степени гармонизированы. Термины «лекарственное растительное сырье» и «лекарственный растительный препарат» во всех рассмотренных документах практически идентичны. Имеются различия в классификации типов ЛРП в документах Европейского союза и США, но принципы определения объемов предоставляемых библиографических и собственных исследований по эффективности и безопасности ЛРП сходны и определяются в первую очередь наличием/отсутствием предшествующего опыта применения препарата у человека, наличием/отсутствием проведенных доклинических и клинических исследований, типом препарата (оригинальный/воспроизведенный), планируемым способом применения (традиционным или с использованием нового пути введения) и показаниями (известные/новые). В регуляторных документах ЕАЭС некоторые из указанных подходов присутствуют лишь частично.
ВЫВОДЫ. Рассмотренные подходы могут быть учтены при создании руководства ЕАЭС по доклиническому и клиническому изучению эффективности и безопасности ЛРП, что позволит обеспечить население препаратами широкого спектра действия, отвечающими современному уровню требований.

ISSN 2619-1164 (Online)
































