


MAIN TOPIC: REASONABLE SUFFICIENCY AS A PRINCIPLE OF PHARMACOTHERAPY

The article discusses the issues of polypharmacy and unjustified prescribing in pregnant women. The author— Honoured Scientist of the Russian Federation, Corresponding Member of the Russian Academy of Sciences, Doctor of Medical Sciences, Full Professor, and Head of the Department for Obstetrics and Gynaecology with a Course on Perinatology at the Institute of Medicine of the Peoples’ Friendship University of Russia named after Patrice Lumumba, Viktor E. Radzinskiy — questions the established practice of unnecessary hospitalisation of pregnant women without clinical indications and prescribing them numerous medicinal products, including antimicrobials, hormones, and antispasmodics. In most cases, pregnancy does not require medical intervention. The widespread use of progestogens in patients with threatened miscarriage lacks an evidence base. Antispasmodics, such as papaverine, are ineffective in threatened preterm labour. The author calls for a rational approach to prescribing medicinal products to pregnant women based on current clinical practice guidelines and the “do no harm” maxim. Viktor E. RADZINSKIY emphasises the need to follow clinical protocols and ensure physicians are informed about the evidence for the use of medicinal products in pregnancy to prevent polypharmacy and adverse consequences.
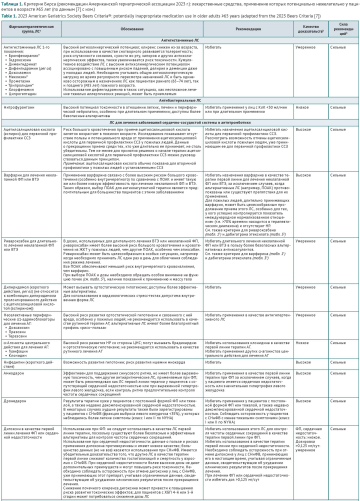
INTRODUCTION. Pharmacotherapy in elderly and senile patients is associated with multimorbidity and polypharmacy and can lead to adverse drug reactions (ADRs). The growth of the world’s population over 60 makes the practical application of the Beers Criteria for Potentially Inappropriate Medication Use in Older Adults, the key recommendations to optimise prescribing in the geriatric population, increasingly important.
AIM. This study aimed to review the history of the Beers criteria, the main changes in the updated 2023 version, and national and international publications on the practical experience of using the criteria in healthcare settings.
DISCUSSION. The criteria for assessing the rationality of pharmacotherapy in older patients were developed by Mark Beers in 1991. His recommendations have been regularly reviewed and updated by a panel of experts from the American Geriatrics Society (AGS); version 7 has been in effect since 2023. The criteria are designed to support pharmacotherapy decision making for adults 65 years old and older in all ambulatory, acute, and institutionalised settings of care, except hospice and end-of-life care settings. The criteria are organised into 5 categories: 1) medications that should be avoided in all older adults; 2) medications considered potentially inappropriate in patients with certain diseases or syndromes; 3) medications that should be used with caution; 4) medications that are potentially inappropriate due to the risk of clinically significant drug–drug interactions; 5) medications that should be avoided or require dosage reduction in patients with impaired renal function. Medicines with pronounced anticholinergic effects are categorised as a separate class. The criteria are based on expert grading of the quality of evidence and strength of recommendations. Compared with the previous version (2019), the updated AGS Beers Criteria® (2023) changed the most in terms of recommendations for anticoagulants and sulfonylureas. According to the results of this review, the AGS Beers Criteria® are actively used in healthcare practice in Russia and abroad to reduce the undesirable effects of potentially inappropriate medication use in elderly patients by optimising the selection of medicines and treatment regimens; to train healthcare providers and patients in the principles of rational pharmacotherapy; to reduce the cost of pharmacotherapy; and to assess the quality of medical care.
CONCLUSIONS. The AGS Beers Criteria® are an effective tool for identifying potentially inappropriate medications in prescribed therapy and selecting appropriate alternatives. Their practical application in healthcare settings can reduce ADRs, hospital admissions, and mortality rates in elderly and senile patients.
INTRODUCTION. The risk of liver damage correlates with the severity of COVID-19. However, a growing number of studies have shown an association between liver function impairment and combinations of medicinal products used to treat COVID-19.
AIM. The study aimed to analyse the annual consumption of medicinal products associated with a high risk of drug-induced liver injury (DILI) used as part of combination therapy in COVID-19 patients and to review a number of medication administration records in order to develop measures to prevent DILI.
MATERIALS AND METHODS. The study used the ATC/DDD methodology to study consumption data for 2020, 2021, and 2022 and analysed a sample of 1250 inpatient medical records and medication administration records of COVID-19 patients treated in a Volgograd region hospital converted into a COVID-19 care centre. For genetically engineered biologicals and cyclophosphamide, which were lacking DDDs, the authors calculated the volume of consumption using the average dose per treatment course. The authors identified medicines capable of causing clinically apparent liver damage (according to the LiverTox database and Russian clinical practice guidelines) and/or elevated liver enzymes in ≥1% of patients (according to safety reports).
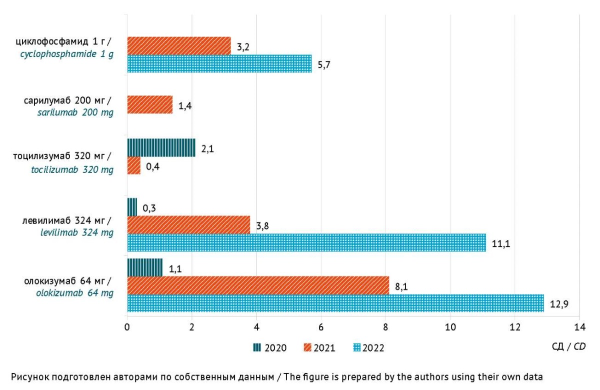
RESULTS. The study found that 28% of the medicinal products used in combination for inpatient treatment of COVID-19 were associated with a high risk of DILI. In 2020, 2021, and 2022, the total consumption of medicinal products associated with a high risk of DILI was 342.3, 425.3, and 402.3 DDDs per 100 bed days, and the total consumption of genetically engineered biologicals (administered as a single dose) and cyclophosphamide was 3.5, 16.9, and 29.7 average course doses per 100 patients, respectively. According to the selective analysis of medical records, 19.8% (247/1250) reported concomitant use of 5 or more medicinal products associated with a high risk of DILI, which increased the risk of adverse drug interactions leading to DILI. In 2022, the most prescribed medicinal products with a high risk of DILI were omeprazole (188.7 DDDs per 100 bed days), non-steroidal anti-inflammatory drugs and paracetamol (54.4 DDDs per 100 bed days), atorvastatin (46.2 DDDs per 100 bed days), levofloxacin (26.4 DDDs per 100 bed days), ceftriaxone (20.5 DDDs per 100 bed days), favipiravir (17.3 DDDs per 100 bed days), and genetically engineered biologicals (24.0 DDDs per 100 bed days).
CONCLUSIONS. To reduce the risk of DILI in COVID-19 patients admitted to infectious disease units, including the risk of DILI due to drug interactions, it is necessary to limit the use of hepatotoxic antibacterial agents, proton-pump inhibitors, and non-steroidal anti-inflammatory drugs, or consider alternative medicinal products with a lower risk of hepatotoxicity.
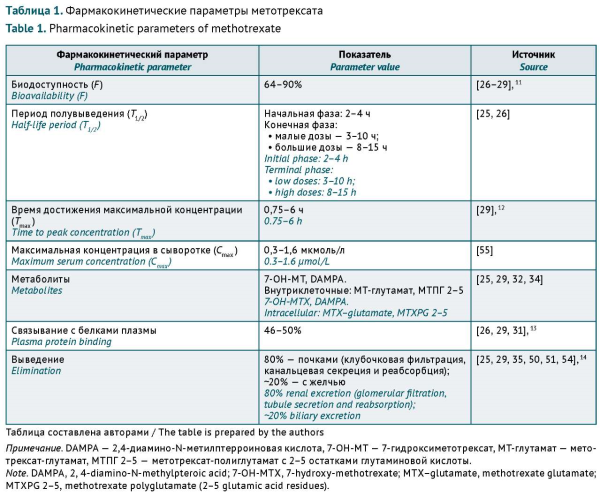
INTRODUCTION. Methotrexate (MTX) is the main disease-modifying antirheumatic drug (DMARD) and the gold standard for the safety and efficacy evaluation of biologicals and targeted small molecules. However, its narrow therapeutic range, interpatient variability in pharmacokinetics and pharmacodynamics, and potential clinically relevant drug–drug interactions (DDIs) may lead to treatment failure and increase the risk of adverse drug reactions (ADRs).
AIM. The study aimed to describe the main clinically significant DDIs associated with MTX used in rheumatic disease therapy and determine possible approaches to addressing this issue based on a literature review.
DISCUSSION. MTX is characterised by pharmacokinetic DDIs during absorption, cell penetration, and elimination. Some non-steroidal anti-inflammatory drugs (NSAIDs), theophylline, sulfasalazine, antibacterial agents, and proton pump inhibitors (PPIs) affect MTX elimination and therapeutic effects. The main ADRs associated with MTX include haematotoxicity, hepatotoxicity, lung tissue damage (interstitial pneumonitis and pulmonary fibrosis), and renal dysfunction. The severity of these ADRs depends on the dose, comorbidities, and concomitant therapy. The toxicity of MTX may be increased by the concomitant administration of medicinal products that exhibit haematotoxicity and affect renal function (impair the elimination of medicines). When co-administering MTX and medicines having clinically significant DDIs described in the literature, healthcare providers should consider the risk factors for each individual patient. The most significant risk factors include moderate to severe renal and hepatic impairment, older age, polypharmacy, and hypoalbuminemia.
CONCLUSIONS. This article describes potential clinically significant interactions between MTX and certain NSAIDs, antibacterial agents, and PPIs that depend on individual patient characteristics and may increase the toxicity or decrease the effectiveness of MTX. MTX deprescribing, short-term withdrawal, and dosing optimisation may be considered as approaches to DDI risk mitigation.
INTRODUCTION. The outbreak of foodborne botulism that occurred in Russia in June 2024 once again demonstrated the danger of this rather rare but severe infectious disease caused by ingesting botulinum neurotoxin. The only aetiological treatment for botulism is currently the administration of antitoxins against various serotypes of botulinum toxin. However, antitoxins do not provide rapid regression of neurological symptoms, which may raise doubts about the effectiveness of the selected treatment option. It is impossible to assess the potential of specific treatment without understanding the mechanisms of action of botulinum toxin and antitoxin.
AIM. This study aimed to systemise information on the mechanism underlying the damaging effect of botulinum neurotoxin, aetiological antitoxin treatment, and the patient recovery process.
DISCUSSION. The mechanism underlying the damaging effect of botulinum neurotoxin consists in the destruction of SNARE proteins in presynaptic cholinergic nerve terminals, which disrupts the release of acetylcholine into the synaptic cleft and the transmission of excitation between neurons. The lack of acetylcholine at the neuromuscular junction results in a distinctive form of persistent flaccid paralysis. The specific mechanism of action of botulinum toxin determines the treatment strategy, which includes a set of life-sustaining measures and the earliest possible antiserum administration. If used within 48 hours from the onset of symptoms, botulinum antitoxin binds botulinum toxin circulating in the blood, which stops the progression of paralysis and prevents further disorders in patients. However, botulinum antitoxin cannot neutralise the effect of the toxin that has already bound to nerve receptors, so clinical symptoms may worsen within 12 hours after antiserum administration. Restoration of normal neuronal transmission occurs through the formation of new axonal sprouts and can take a long time.
CONCLUSIONS. Antitoxin administration is effective and irreplaceable in the aetiological treatment of botulism. Nevertheless, the duration of recovery depends on the speed of reinnervation and restoration of transmission at the neuromuscular junction.
PHARMACOVIGILANCE
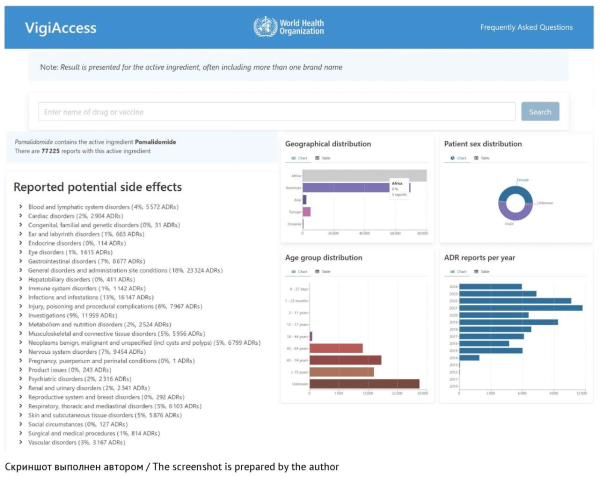
INTRODUCTION. Postmarketing surveillance is the main mechanism to monitor and evaluate the safety of drugs approved for widespread clinical use. This mechanism provides up-to-date information on adverse drug reactions and facilitates the implementation of necessary measures to prevent or minimize the risks associated with pharmacotherapy. Marketing authorization holders should regularly conduct searches for and analyses of drug safety data from all available sources. The lack of practical recommendations for selecting relevant information sources complicates the development of an optimal strategy for collecting drug safety data necessary for timely detection of changes in the safety profile of a drug that may affect the benefit–risk ratio.
AIM. This study aimed to select the most appropriate methods for collecting drug safety information from the open sources used to monitor the safety profiles of approved drugs and assess their benefit–risk ratios.
DISCUSSION. The main sources of new information on the safety of drugs include pharmacovigilance databases, websites of regulatory authorities, publications in medical scientific journals, and real-world clinical practice. According to the analysis results, the most widely used databases are the World Health Organization (WHO) VigiBase database, the European Union (EU) EudraVigilance database, and the United States Food and Drug Administration (FDA) Adverse Event Reporting System (FAERS). This article also discusses the capabilities and interfaces of various applications for working with safety data, as well as the conditions for accessing databases. Additionally, the article provides detailed instructions on how to search for safety information on the websites of the European Medicines Agency (EMA) and FDA, which are considered to be the most reliable sources of information. Further, the article provides an overview of reputable medical journals most likely to publish articles on adverse drug reactions. In addition, the article covers bibliographic databases and search engines, which can simplify the search for scientific publications. Systematic monitoring of these sources can help marketing authorization holders to effectively assess the safety profiles and benefit–risk ratios of approved drugs.
CONCLUSIONS. The use of recommended data sources can optimize the process of safety monitoring, significantly increase the identification rate for potential risks of pharmacotherapy, and facilitate the timely development of measures to prevent these risks. This, in turn, can contribute to improving the safety of patients and the quality of medical care.
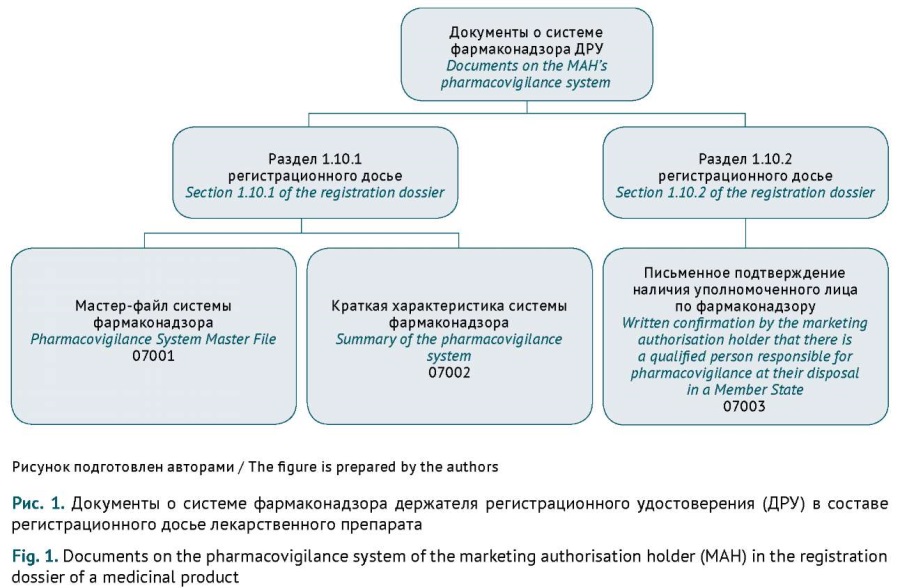
INTRODUCTION. Documents on the pharmacovigilance system of the marketing authorisation holder (MAH), including the Pharmacovigilance System Master File (PSMF) and the PSMF-based summary of the pharmacovigilance system (SPS), are a mandatory component of the registration dossier for a medicinal product. Applicants must submit and update these documents in strict accordance with the legislation of the Eurasian Economic Union (EAEU). Systematisation of the requirements for drafting and submitting documents on the pharmacovigilance system will help applicants streamline the documentation process.
AIM. This study aimed to analyse the requirements for submitting either the PSMF or the SPS, depending on the EAEU marketing authorisation procedure, and to describe typical errors made by applicants when submitting these documents.
DISCUSSION. The content of dossier documents on the pharmacovigilance system is regulated by the Rules of Good Pharmacovigilance Practice of the Eurasian Economic Union (Decision No. 87 of the Council of the Eurasian Economic Commission (EEC) dated 3 November 2016), and their submission is governed by the Rules of Marketing Authorisation and Assessment of Medicinal Products for Human Use (EEC Council Decision No. 78 dated 3 November 2016). MAHs are required to keep documents on the pharmacovigilance system and the corresponding registration dossiers up to date. This article summarises the specific requirements for submitting either the PSMF or the SPS, depending on the EAEU marketing authorisation procedure. Additionally, this article highlights typical errors made by MAHs when preparing documents on the pharmacovigilance system. According to the EAEU Rules of Good Pharmacovigilance Practice, the first application submitted for marketing authorisation of a medicinal product in the EAEU should include the PSMF, and subsequent applications should include the SPS as part of the registration dossier. Changes to the pharmacovigilance documents should be made in accordance with the classifier (EEC Council Decision No. 78 dated 3 November 2016).
CONCLUSIONS. This analysis of the requirements for the PSMF or the SPS as part of various marketing authorisation procedures will facilitate compliance with the requirements of the EAEU legislative acts and ensure correct submission of necessary documents on the pharmacovigilance system by applicants. In addition, regulatory authorities will make fewer requests to submit missing information and grant more marketing authorisations.
ERRATUM
Erratum: Velts N.Yu. et al. Submission of Documents on the Pharmacovigilance System as Part of the Registration Dossier within the Framework of the EAEU Procedures: Analysis of Requirements and Typical Errors
https://www.risksafety.ru/jour/article/view/476
PRECLINICAL STUDIES
INTRODUCTION. Fluoroquinolones are antibacterial agents associated with adverse drug reactions (ARDs), including aortic lesions; this ARD risk limits the use of fluoroquinolones. Moreover, fluoroquinolones have been reported to induce lesions in other connective tissues (cartilage, tendons), associated with magnesium deficiency.
AIM. The study aimed to analyse the effects of magnesium orotate on the thoracic aorta in laboratory rabbits treated with levofloxacin.
MATERIALS AND METHODS. The study randomised laboratory rabbits into 3 groups of 10 animals each to receive oral doses of either the carrier solution (control group), or 150 mg/kg/day levofloxacin (levofloxacin group), or 150 mg/ kg/day levofloxacin and 140 mg/kg/day magnesium orotate (levofloxacin/magnesium group). After 14 days of treatment, venous blood samples were taken to determine the serum levels of magnesium, matrix metalloproteinase-9 (MMP-9), and tissue inhibitor of matrix metalloproteinase-1 (TIMP-1), as well as MMP-9 to TIMP-1 ratios. The authors conducted morphological and mechanical characterisation of thoracic aorta samples; the mechanical characterisation involved uniaxial tensile testing. Data are presented as the mean and standard deviation values.
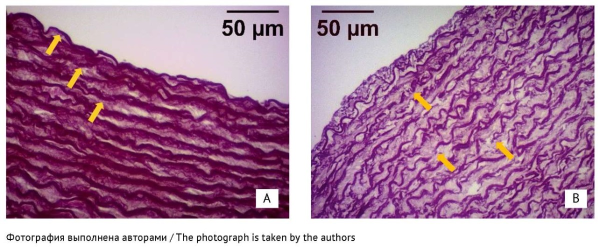
RESULTS. The study did not detect any changes in the serum MMP-9, TIMP-1, and magnesium levels or in the MMP-9/TIMP-1 ratios. The authors identified foci of moderate elastic fibre fragmentation in the aortic media in 5 of 10 aortic samples from the levofloxacin group, in 1 of 10 samples from the levofloxacin/magnesium group, and in none from the control group (p=0.013). Rabbits from the levofloxacin group had significantly fewer medial elastic membranes than the others (p=0.015; vs the control group: p=0.022), and their elastic mem
QUALITY OF MEDICINES
INTRODUCTION. This work continues the authors’ earlier study on gelatine capsule contamination with elemental impurities, which resulted in proposing a list of elements for standardisation. To ensure the safe use of medicinal products in this dosage form, it is necessary to develop criteria for the quantitative assessment of potential human health risks associated with elemental impurities in gelatine capsule shells.
AIM. This study aimed to determine the permitted daily concentration (PDC) limits for elemental impurities in gelatine capsules from a risk-based pharmaceutical quality control perspective.
MATERIALS AND METHODS. The authors analysed their previous experimental data, the relevant Russian State Standard (GOST), the requirements of the State Pharmacopoeia of the Russian Federation and leading world pharmacopoeias (USA, UK, EU, China, Japan, and India), and the scientific literature published from 1980 to 2023 on the content of elemental impurities (Al, As, Cd, Cu, Cr, Cr, Fe, Hg, Mn, Pb, and Zn) in gelatine capsule shells and medical gelatine.
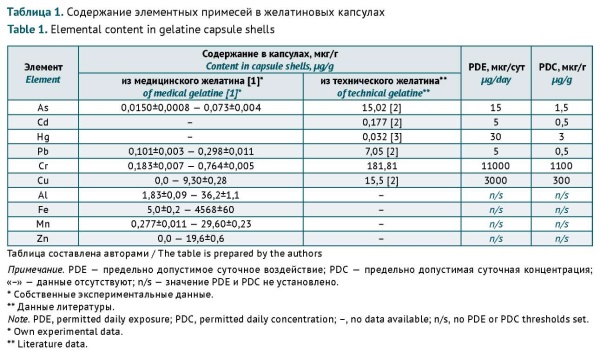
RESULTS. When standardising the content of elemental impurities in gelatine capsule shells, it is necessary to consider not only the toxicity of impurities but also their potential to compromise the quality of gelatine. Complexing elements (Cd, Cu, Hg, Cr, and Zn) can affect the quality of gelatine at subtoxic concentrations. For Cd, Cu, and Hg, the authors recommended using the PDC limits specified in GOST 23058-89 Gelatine. Raw materials for the medical industry. Specifications. For Cr and Zn, the authors recommended adhering to the PDC limits specified in the State Pharmacopoeia of the Russian Federation, edition XIV, Monograph 2.1.0099.18 Gelatine. Lead affects the quality of gelatine at concentrations above the toxicity threshold, and arsenic does not cause clustering of gelatine molecules. For Pb and As, the authors recommended using the PDC limits specified in the State Pharmacopoeia of the Russian Federation, edition XV, General Monograph 1.1.0040 Elemental impurities. The authors proposed a conversion factor to account for the complexing potential of Al when limiting its content in gelatine capsule shells. The authors recommended assessing the risks to human health associated with Mn intake with gelatine capsule shells indirectly by listing total heavy metals as a quality attribute to be controlled at the level of medical gelatine. The content of Fe should also be standardised at the raw material level.
CONCLUSIONS. The content of elemental impurities in gelatine capsule shells should not exceed the following limits: 45 µg/g for Al, 1.5 µg/g for As, 0.03 µg/g for Cd, 10 µg/g for Cr, 15 µg/g for Cu, 0.05 µg/g for Hg, 0.5 µg/g for Pb, and 30 µg/g for Zn. The standardisation of these elements in gelatine capsule shells will prevent negative effects on the human body and shell quality.
COCHRANE PUBLICATIONS
This article is the Russian translation of the Plain Language Summary (PLS) of the Cochrane Review previously published in the Cochrane Database of Systematic Reviews. Original publication: Palacios C, Kostiuk LL, Cuthbert A, Weeks J. Vitamin D supplementation for women during pregnancy. Cochrane Database Syst Rev. 2024;7(7):CD008873. https://doi.org/10.1002/14651858.CD008873.pub5

ISSN 2619-1164 (Online)






























