


MAIN TOPIC: INNOVATIONS AND CHALLENGES IN PHARMACOTHERAPY: FROM DEVELOPING NOVEL MEDICINAL PRODUCTS TO ENSURING THEIR CLINICAL SAFETY
INTRODUCTION. Alzheimer’s disease (AD), which becomes more prevalent with increasing life expectancy, is a leading cause of severe cognitive disorders and dementia. In 2021–2024, the Food and Drug Administration (FDA) approved the first disease-modifying therapies (DMTs) based on anti-amyloid monoclonal antibodies (anti-Aβ mAbs), including aducanumab (accelerated approval), lecanemab, and donanemab. Ongoing studies are evaluating the efficacy and safety of these anti-Aβ mAbs.
AIM. This review aimed to examine the prospects and limitations of anti-amyloid DMTs for AD in the context of the current understanding of AD pathogenesis mechanisms.
DISCUSSION. According to current concepts, the pathogenesis of AD is primarily driven by the aggregation of amyloid plaques and neurofibrillary tangles of hyperphosphorylated tau protein in the brain, which is accompanied by neurodegenerative changes. The pathogenesis of AD is still being studied. The mechanism of action of FDA-approved anti-Aβ mAbs for the treatment of AD (aducanumab, lecanemab, and donanemab) involves microglial activation followed by amyloid phagocytosis and degradation. The mAbs differ in their affinity to different amyloid species. Clinical trials (average duration: 1.5 years) have demonstrated that all 3 anti-Aβ mAbs reliably and significantly reduce the brain amyloid burden (up to complete amyloid clearance with donanemab) and slow down cognitive decline in patients with early-stage AD. Although reliable, the reduction in cognitive decline rates is of limited clinical significance. The most common adverse event of mAb therapy is amyloid-associated imaging abnormalities (ARIA) observed in 20–30% of patients. This complication is a result of amyloid clearance and typically occurs early in the course of treatment. APOE ε4 allele carriers have a higher incidence of ARIA than non-carriers. Most reported cases of ARIA were asymptomatic and resolved over time.
CONCLUSIONS. Anti-Aβ mAbs have shown reliable efficacy in reducing the brain amyloid burden and slowing the progression of cognitive decline in AD. However, the widespread use of anti-Aβ mAbs has been hampered by their limited clinical efficacy, invasiveness of diagnosis, high diagnostic and treatment costs, and additional expenses associated with adverse event monitoring.
INTRODUCTION. Vaccination against smallpox was discontinued in 1980 following the success of the World Health Organisation’s Smallpox Eradication Programme. Consequently, more than half of the current world population lacks immunity to orthopoxviruses pathogenic to humans. Since 2022, several countries have been reporting outbreaks of mpox (previously known as monkeypox), which necessitates developing new small molecules active against orthopoxviruses.
AIM. The aim was to evaluate the safety and tolerability of NIOCH-14, a Russian anti-smallpox medicinal product, after oral administration in a phase I clinical trial.
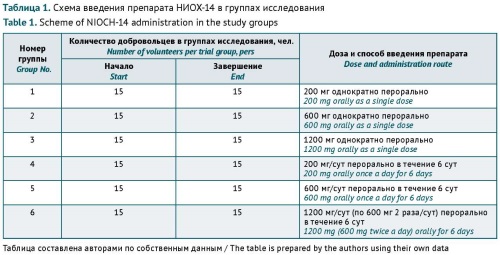
MATERIALS AND METHODS. The clinical trial was conducted in 90 healthy volunteers at the Federal State Budgetary Institution of Healthcare ‘Medical and Sanitary Unit No. 163 of the Federal Medical and Biological Agency’ in accordance with Protocol No. NIOCH-01/20 ‘An open, simple, randomised study of the safety, tolerability, pharmacokinetics of NIOCH-14 in volunteers aged 18–50 years in parallel groups’. Investigators used a standard set of clinical, laboratory, and instrument-based testing methods. The clinical trial included six experimental groups of 15 subjects each. Groups 1, 2, and 3 received NIOCH-14 as a single oral dose of 200, 600, and 1200 mg, respectively. Groups 4, 5, and 6 received NIOCH-14 at daily oral doses of 200, 600, and 1200 mg/day, respectively, for 6 days.
RESULTS. After single and repeated administration of NIOCH-14 at doses of 200, 600, and 1200 mg to volunteers, the study did not demonstrate any changes in the blood test results (erythrocyte, leucocyte, platelet, and differential leucocyte counts; haemoglobin levels; erythrocyte sedimentation rate values; alanine transaminase, aspartate transaminase, and alkaline phosphatase activity levels; total protein, total bilirubin, glucose, creatinine, urea, and C-reactive protein levels; prothrombin time ratios; thymol turbidity). Similarly, the urinalysis results remained unaltered throughout the entire observation period. No adverse events were observed in volunteers receiving NIOCH-14 at all single doses or repeated doses of 200 and 600 mg. One volunteer receiving the investigational medicinal product at a repeated dose of 1200 mg/day experienced transient epigastric pain on days 2 and 5.
CONCLUSIONS. The physical examination, haematology, blood chemistry, and urinalysis results did not reveal any pathological changes in healthy volunteers. The investigational medicinal product, formulated as hard gelatin capsules containing 200 mg of NIOCH-14 and excipients, demonstrated a favourable safety and tolerability profile.

INTRODUCTION. The use of relevant species of laboratory animals in preclinical safety studies during the development of novel medicines provides valuable information for assessing the risks and benefits of such medicines for humans. The appropriate species are selected upon consideration of scientific, ethical, and practical aspects, and the choice should be justified. Regulatory documents of the Eurasian Economic Union (EAEU) indicate that preclinical safety studies of medicines should use relevant species of animals, but the recommendations for their choice are insufficient. Therefore, it is essential to analyse information from international regulatory documents on preclinical studies and recommendations from the scientific community to identify meaningful criteria that can be used to select experimental animals for preclinical studies.
AIM. This study aimed to analyse the current regulatory, scientific, and methodological framework in order to identify key factors and criteria for substantiating the choice of relevant species of experimental animals for preclinical safety studies.
DISCUSSION. This article analyses guidelines on preclinical studies issued by the EAEU, the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH), and the European Medicines Agency (EMA), as well as scientific publications on selecting experimental animals. The findings suggest that the most comprehensive recommendations for selecting relevant animals are provided in the EAEU Rules for conducting studies of biological medicinal products as well as the ICH S6(R1) guideline, the ICH S5(R3) guideline on reproductive toxicity studies, the ICH S11 guideline on the development of paediatric pharmaceuticals, and the EMA guideline on strategies to identify and mitigate risks of the first-in-human use of medicinal products. Selecting suitable animals for preclinical studies has been a subject of lively scientific debate. According to research sponsors, the most common regulatory requests related to animal relevance are to provide additional information on the pharmacological relevance of the selected species, to justify the use of only one species, or to conduct additional studies in other species. Many research teams use internal documents that describe the stages and criteria that facilitate the selection of relevant experimental animals. The scientific community has offered over 40 different parameters that, when assessed in vitro, in vivo, and in silico, can help researchers justify the relevance of experimental animals for preclinical safety studies.
CONCLUSION. Selecting relevant test systems and models for preclinical safety studies is a scientific endeavour in its own right. To justify the relevance of experimental animals, ensure the translatability of results, and comply with ethics principles, the most valuable criteria are the criteria developed using a systemic approach based on in vitro and in vivo analysis of a set of pharmacodynamic, pharmacokinetic, and toxicological parameters.
INTRODUCTION. Newly identified risks associated with the use of fluoroquinolones and the spread of antimicrobial resistance make the identification and analysis of medication errors (MEs) in prescribing fluoroquinolones especially important for providing rational antibiotic therapy. Fluoroquinolones that are most commonly used in real-world clinical settings include levofloxacin.
AIM. This study aimed to examine the pattern of MEs associated with fluoroquinolones, exemplified by levofloxacin, through an analysis of spontaneous reports (SRs) submitted to the Russian pharmacovigilance database.
MATERIALS AND METHODS. The authors retrospectively analysed the SRs of adverse drug reactions (ADRs) submitted to the Russian pharmacovigilance database between 1 April 2019 and 28 February 2023. According to the selected inclusion criteria, the study focused on the SRs that specified levofloxacin as a suspect medicinal product and described ADRs that took place in the Russian Federation. ME identification used summaries of product characteristics for levofloxacin approved in Russia, official clinical guidelines, and the antimicrobial stewardship guidelines Strategy for the Control of Antimicrobial Therapy (SCAT).
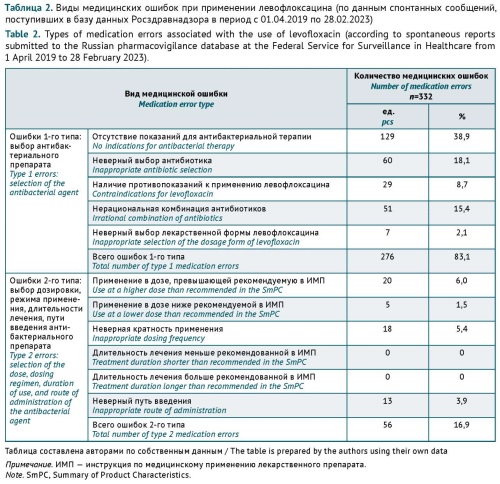
RESULTS. The analysis included 950 SRs. MEs were identified in 307 (32.3%) of these SRs, and the total number of MEs was 332. MEs associated with the selection of the medicinal product included prescribing levofloxacin to patients without an indication for antibacterial therapy (38.9%, n=129, with 76.0% of cases being viral infections), incorrect selection of levofloxacin as a first-line antibacterial agent (18.1%, n=60), and irrational and excessive prescribing of levofloxacin in combination with other antibacterial agents (15.4%, n=51). Less frequently identified MEs were related to inappropriate dosing (13.0%, n=43), levofloxacin use in patients with contraindications (8.7%, n=29), and incorrect selection of the route of administration (3.9%, n=13) and the dosage form (2.1%, n=7).
CONCLUSIONS. According to the results of this study, the practice of prescribing antibacterial agents for viral infections persists despite strong evidence of ineffectiveness in such cases. Antibacterial agents can be used effectively and safely only if prescribed for approved indications, administered at recommended doses, and delivered via specified routes of administration. The overuse of antibiotics may have negative sequelae not only for the health of an individual patient but for the health of the general population because of the increased risk of antimicrobial resistance. Therefore, there is a need to develop measures to limit the excessive use of antibiotics.
INTRODUCTION. Drug-induced liver injury (DILI) is a common and potentially life-threatening complication associated with the use of medicinal products and bioactive dietary supplements. Recognising early signs of liver damage can be challenging for a number of reasons, including polypharmacy, comorbidity, pre-existing liver conditions, and misinterpretation of non-specific clinical manifestations. It is necessary to consolidate information on the diagnostic and prognostic options for DILI that are available to physicians in outpatient settings.
AIM. This study aimed to provide consolidated and systematised information on the risk factors, early diagnosis methods, and prognostic assessment methods for DILI to provide recommendations for DILI identification in outpatient settings.
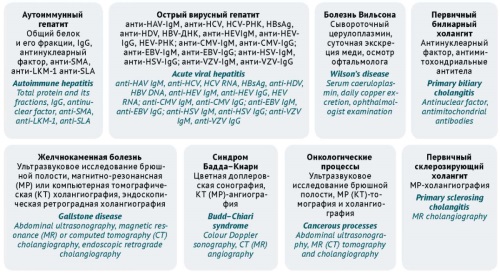
DISCUSSION. The main risk factors that contribute to the development of DILI are potentially hepatotoxic medicinal products, high doses of medicinal products, polypharmacy, a history of liver disease, and genetic predisposition. Even though DILI can occur with any medicinal product, outpatient physicians should be aware of the medicinal products that are most often associated with this condition. The initial screening for liver injury and the determination of its severity necessitate taking a comprehensive medication history and conducting standard liver function tests (alanine transaminase, aspartate transaminase, alkaline phosphatase, total bilirubin). Tools relevant for outpatient care include scales that help identify the cause-and-effect relationship between a medicinal product and liver damage. These include the Council for International Organisations of Medical Sciences/Roussel Uclaf Causality Assessment Method (CIOMS/RUCAM) scale, the Maria & Victorino scale (M&V), the Digestive Disease Week–Japan (DDW–J) scale, the Naranjo Adverse Drug Reaction Probability Scale). The CIOMS/RUCAM scale is suitable for suspected hepatocellular or cholestatic DILI, and the M&V scale is additionally adapted for mixed DILI and includes points for extrahepatic manifestations. Other relevant tools for outpatient care are those for predicting the severity of DILI (Hy’s law).
CONCLUSIONS. Outpatient physicians should adopt an approach that combines medication history collection, physical examination, laboratory investigations, and instrument-based diagnostics with the routine use of scales for establishing causality between a medicinal product and DILI. This approach will help predict the clinical course of DILI at an early stage and make a quick decision on further treatment.
INTRODUCTION. Antipsychotic-induced parkinsonism (AIP) is an extrapyramidal adverse drug reaction (ADR) associated with antipsychotics (APs). Despite its classification as a non-serious ADR, AIP significantly decreases the quality of life in patients with schizophrenia spectrum disorders, which makes early diagnosis and timely management of AIP an urgent issue.
AIM. This study aimed to develop a risk assessment scale and a personalised diagnostic algorithm for AIP as the most common and clinically significant neurological ADR in patients with schizophrenia spectrum disorders.
MATERIALS AND METHODS. The authors analysed modifiable and non-modifiable risk factors for AIP, as well as rating scales, questionnaires, and laboratory testing methods to diagnose the condition. The analysis was based on full-text publications in Russian or in English sourced from the eLIBRARY.RU, PubMed, Springer, ClinicalKey, and Google Scholar databases. As a preliminary step, the authors compared the effectiveness of validated AIP risk assessment scales, including the Simpson–Angus Scale (SAS), the Extrapyramidal Symptom Rating Scale (ESRS), the Unified Parkinson’s Disease Rating Scale (UPDRS), the Hoehn and Yahr scale (H&Y Scale), the Webster Rating Scale, and the Mindham Rating Scale. Comparisons were made regarding the duration of testing, the degree of reliability in assessing clinical manifestations of AIP, and the ability to assess risk factors (predictors) of AIP and the rate of AIP development. The results obtained formed the basis for developing an AIP riskometer and a diagnostic algorithm.
RESULTS. The authors developed an original risk assessment scale for diagnosing and predicting AIP. Directions for personalised patient management were determined for patients at high and medium risk of AIP. This article presents an algorithm for diagnosing AIP in patients with schizophrenia spectrum disorders in two variants based on pro-reactive (predictive) or reactive pharmacogenetic testing. According to the study results, pro-reactive pharmacogenetic testing can help determine the risk of AIP in a patient before primary therapy.
CONCLUSIONS. The risk assessment scale and the personalised diagnostic algorithm developed by the authors may be useful for practising neurologists, psychiatrists, and clinical pharmacologists. The development and clinical implementation of novel tools for risk assessment, prevention, and diagnosis of AIP—the most common AP-associated neurological ADR—can improve the quality of treatment and preventive care for patients with schizophrenia spectrum disorders.
INTRODUCTION. Studying the effect of food on the bioavailability of medicinal products is important for selecting the right administration conditions for generics (in bioavailability and bioequivalence studies) and confirming the selection for originators in different patient groups (in clinical trials). However, there are currently no common harmonised requirements for food-effect bioavailability studies.
AIM. This study aimed to evaluate the conditions for investigating the effect of food on the bioavailability of medicinal products in bioequivalence studies through an analysis of the national and international regulatory requirements
for the conduct of clinical bioequivalence studies; additionally, this study aimed to identify common and unique requirements applied in different countries with a view to selecting the optimal conditions for conducting
bioequivalence studies of medicinal products.
DISCUSSION. Food-effect bioavailability studies of medicinal products should rely on the Biopharmaceutics Classification System (BCS) and the Biopharmaceutical Drug Disposition and Classification System (BDDCS), which
classify medicinal products by solubility, permeability, and metabolism. This study analysed documents reflecting the approaches of international organisations to bioequivalence studies, including documents by the World
Health Organisation (WHO), the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH), and regulatory bodies of the Eurasian Economic Union, the European Union
(European Medicines Agency (EMA)), and the United States of America (Food and Drug Administration (FDA)). The analysis revealed differences in the conditions for studying the effect of food on the bioavailability of medicinal
products. A common approach is to require that bioequivalence studies should be conducted under standardised conditions. The differences lie in the expected scope of postprandial studies; the recommended meal
composition with regard to the energy, protein, carbohydrate, and fat content and local dietary preferences; and approaches to food-effect bioavailability studies of high-risk medicinal products, medicinal products with
linear and non-linear pharmacokinetics, and modified-release formulations.
CONCLUSIONS. The differences identified in the national and international requirements for the conduct of food-effect bioavailability studies of medicinal products underscore the need for regulatory standard harmonisation, which will contribute to ensuring the safe and effective use of medicinal products, to implementing uniform approaches to the interpretation of the results of food-effect bioavailability studies, and to bringing medicinal products to the global pharmaceutical market.
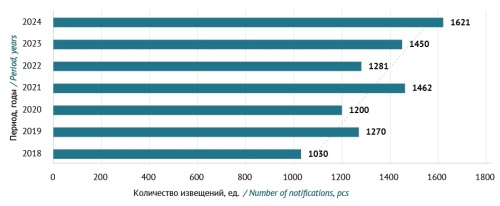
INTRODUCTION. It is essential to monitor the efficacy and safety of medicinal products as part of post-marketing surveillance to identify, assess, and prevent adverse drug reactions (ADRs). The effectiveness of pharmacovigilance depends largely on healthcare professionals’ adherence to the requirements and timeframes for reporting information to regulatory authorities.
AIM. This study aimed to identify and systematise the key aspects of pharmacovigilance through an analysis of experience in organising pharmacovigilance in Moscow-based healthcare institutions in 2018–2024 to determine the focus areas for implementing, monitoring, and improving the pharmacovigilance system operating in medical organisations.
DISCUSSION. The significance of pharmacovigilance systems and the need for their continuous improvement are underpinned by experience in establishing pharmacovigilance databases, including the global VigiBase database, the European Union’s EudraVigilance database, and the Eurasian Economic Union’s database. The main pharmacovigilance tools include active surveillance and collection of unsolicited/solicited reports of suspected ADRs. The Russian Federal Service for Surveillance in Healthcare (Roszdravnadzor) has developed clear operational criteria for pharmacovigilance in medical organisations. These criteria require that medical organisations should have in-house regulations for collecting, registering, and reporting data on ADRs to Roszdravnadzor, appointing qualified persons responsible for pharmacovigilance, and obtaining access to the Pharmacovigilance database of Roszdravnadzor’s Automated Information System. This article provides a detailed description of tools for establishing an effective pharmacovigilance system in a medical organisation, as well as the focus areas for organising pharmacovigilance in medical organisations, identified by analysing the experience of a territorial entity of the Russian Federation.
CONCLUSIONS. To improve the effectiveness of pharmacovigilance in medical organisations, it is necessary to ensure the quality, integrity, and completeness of data submitted in spontaneous ADR reports. Spontaneous reporting should be supplemented with active surveillance methods, including the Global Trigger Tool. The current pharmacovigilance system in Moscow demonstrates a steady increase in reporting ADRs to Roszdravnadzor.
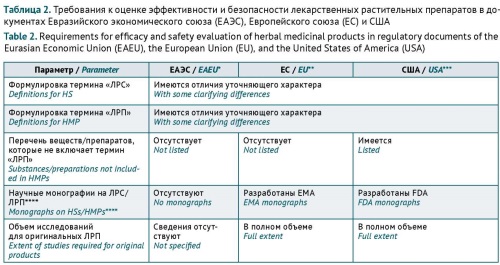
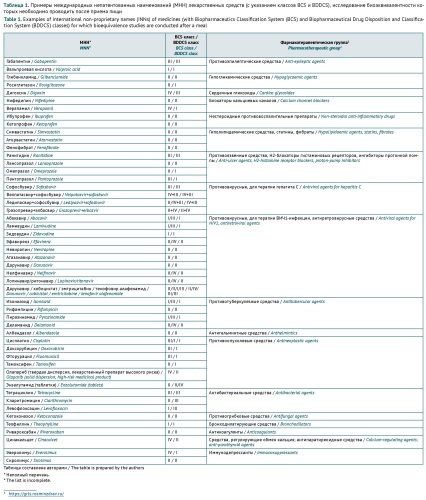
INTRODUCTION. Herbal medicinal products are widely used in medical practice. Special considerations apply to the extent of safety and efficacy studies required for herbal medicinal products in different countries, as documented in their marketing authorisation frameworks. Currently, the Eurasian Economic Union (EAEU) lacks guidelines on the extent of preclinical and clinical studies required for herbal medicinal products.
AIM. This study aimed to analyse the possibility of using international standards and approaches in the development of the EAEU guidelines for preclinical and clinical studies of the safety and efficacy of herbal medicinal products.
DISCUSSION. First of all, marketing authorisation of herbal medicinal products involves special considerations because these medicinal products contain complex mixtures of bioactive substances. According to the analysis of the regulatory approaches of the European Union (EU), the United States of America (USA), and the EAEU, the safety and efficacy testing requirements for herbal medicinal products are harmonised to a certain degree. The terms used for herbal substances and herbal medicinal products have almost identical definitions in all the studied documents. Despite the differences in their typological classifications of herbal medicinal products, the EU and USA documents provide similar principles for determining the required extent of published data and original studies on the safety and efficacy of herbal medicinal products. Mainly, the extent depends on the herbal medicinal product’s history of previous human use and completed preclinical and clinical studies (if any), type (original/generic), intended administration route (traditional/new), and indications (established/new). Some of the approaches presented in the article are only partially included in the current EAEU regulatory documents.
CONCLUSIONS. The discussed approaches can be considered in the development of the EAEU guidelines for preclinical and clinical studies of the safety and efficacy of herbal medicinal products. Such guidelines will contribute to providing the population with broad-spectrum herbal medicinal products that meet current safety and efficacy standards.

ISSN 2619-1164 (Online)






























